XiaoMi-AI文件搜索系统
World File Search SystemSNV

将个性化药物带到新的水平
BostonGene achieves the most comprehensive overview of a tumor through whole exome (WES) and transcriptome (RNAseq) sequencing to detect genomic alterations such as fusions, single nucleotide variants (SNV), indels, copy number alterations (CNA), tumor mutational burden (TMB), microsatellite instability (MSI), fusions, frameshifts,超过20,000个基因的重排和表达水平。Bostongene肿瘤肖像™测试结合了精确的数据处理和专有算法,以生成易于理解的肿瘤示意图,即分子功能肖像*。

非线性量子光子量,带有锡面中心与一维钻石波导
单光子和固态颜色中心之间的非线性相互作用是量子科学中许多应用的核心[1,2],例如实现量子互联网[3,4]。尤其是,钻石中的彩色中心已启用了这个方向的高级演示,显示了多键量子网络操作[5,6],内存增强的通信[7]和可扩展的芯片载荷混合动力集成[8]。Among the diamond color centers, the tin-vacancy center (SnV) has recently emerged as a promising qubit platform, as it combines the inversion symmetry of group-IV color centers [9,10] , allowing for integration in nanophotonic structures, with good optical properties [11 – 14] and above-millisecond spin coherence at temperatures above 1 K [15,16] .将光子整合与自旋和光学控制结合的设备可以用作实现自旋photon大门的未来可伸缩构建块[17]。在通往这种可扩展的片上整合的路径上,将发射剂掺入纳米光子波导中[12,18],可以探索相干的发射极 - 光子相互作用,典型的波导 - 耦合系统[19,20]。与纳米光腔相比[21],波导具有宽带的优势,消除了腔体调整的挑战,并且在制造中具有明显更高的误差耐受性。 在这封信中,我们提出了一个由SNV中心组成的设备,该中心与纳米型钻石波导搭配锥形纤维通道,如图所示 1(a)。 感谢有效的耦合,双面访问和实时与纳米光腔相比[21],波导具有宽带的优势,消除了腔体调整的挑战,并且在制造中具有明显更高的误差耐受性。在这封信中,我们提出了一个由SNV中心组成的设备,该中心与纳米型钻石波导搭配锥形纤维通道,如图1(a)。感谢有效的耦合,双面访问和实时

使用 ACEofBASEs 靶标预测对具有碱基编辑的 DNA 变异进行精确的体内功能分析
摘要单核苷酸变异 (SNV) 是影响个体性状和疾病易感性的普遍遗传因素。碱基编辑器、橡胶和铅笔基因组编辑工具的最新开发和优化现在有望实现对模型生物中的 SNV 进行直接功能评估。然而,缺乏有助于靶标预测的生物信息学工具限制了碱基编辑在体内的应用。在这里,我们为青鳉 (Oryzias latipes) 和斑马鱼 (Danio rerio) 中的腺嘌呤和胞嘧啶碱基编辑提供了一个框架,非常适合可扩展的验证研究。我们开发了一个在线碱基编辑工具 ACEofBASEs(对碱基编辑的仔细评估),通过简化 sgRNA 设计和进行脱靶评估来促进决策。我们在青鳉和斑马鱼中使用最先进的腺嘌呤 (ABE) 和胞嘧啶碱基编辑器 (CBE) 来高效编辑眼色素沉着基因和转基因 GFP 功能。编码肌钙蛋白 T 和钾通道 ERG 的基因中的碱基编辑忠实地再现了已知的心脏表型。等位基因的深度测序揭示了预期编辑的丰富性,而 ABE8e 和 evoBE4max 的插入或删除 (indel) 事件水平较低。我们最终在 F0 和 F1 中验证了先天性心脏病 (CHD) dapk3、ube2b、usp44 和 ptpn11 的新候选基因中的错义突变,这些目标基因中有基因型-表型相关性。该碱基编辑框架适用于鱼类中可获得的多种 SNV 易感性状,有助于直接验证候选基因并确定其优先级,以便进行详细的机制下游研究。

大规模基因组和转录组测序分析揭示了植物中高活性腺嘌呤碱基编辑器诱导的突变景观
摘要背景:利用最近开发的 tRNA 腺苷脱氨酶 (TadA8e 和 TadA9) 改造的高活性腺嘌呤碱基编辑器 (ABE) 表现出强大的碱基编辑活性,但引发了人们对脱靶效应的担忧。结果:在本研究中,我们对 ABE8e 和 ABE9 诱导的水稻 DNA 和 RNA 突变进行了全面评估。对用四种 ABE(包括 SpCas9n-TadA8e、SpCas9n-TadA9、SpCas9n-NG-TadA8e 和 SpCas9n-NG-TadA9)转化的植物进行全基因组测序分析表明,含有 TadA9 的 ABE 导致更多数量的脱靶 A 到 G (A>G) 单核苷酸变体 (SNV),而含有 CRISPR/SpCas9n-NG 的 ABE 导致水稻基因组中脱靶 SNV 总数更高。对携带 ABE 的 T-DNA 的分析表明,在 T-DNA 整合到植物基因组之前和/或之后可以引入靶向突变,在 ABE 整合到基因组之后会形成更多的脱靶 A>G SNV。此外,我们在 ABE 表达高的植物中检测到脱靶 A>G RNA 突变,但在 ABE 表达低的植物中未检测到。脱靶 A>G RNA 突变倾向于聚集,而脱靶 A>G DNA 突变很少聚集。结论:我们的研究结果表明 Cas 蛋白、TadA 变体、ABE 的时间表达和 ABE 的表达水平对水稻中的 ABE 特异性有影响,这为了解 ABE 的特异性提供了见解,并提出了除改造 TadA 变体之外增加 ABE 特异性的其他方法。
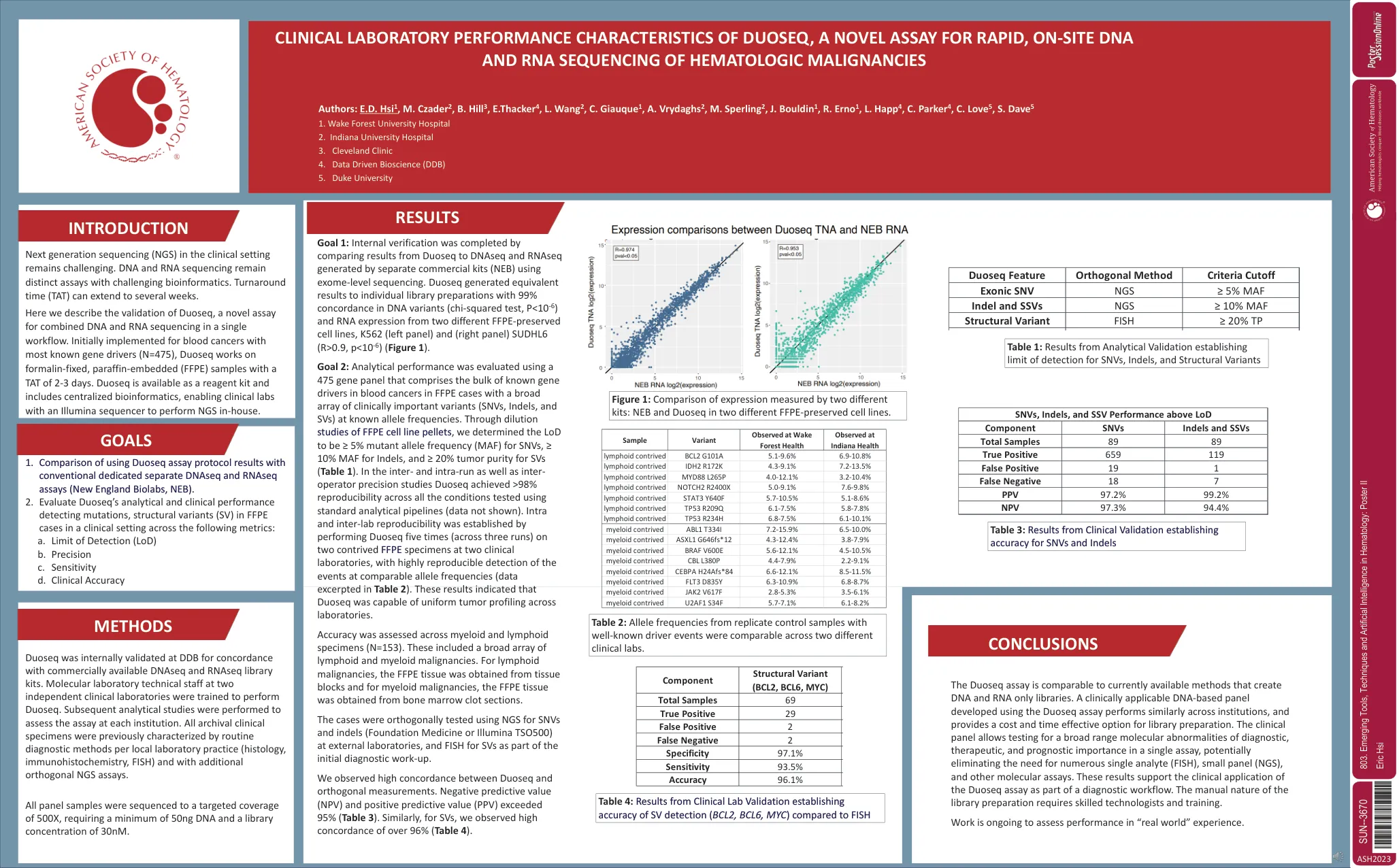
Duoseq的临床实验室绩效特征,A ...
目标2:使用475个基因面板评估了分析性能,该基因面板包括在已知等位基因频率下具有广泛临床重要变体(SNV,INDELS和SVS)的FFPE病例中大量已知基因驱动因素。通过对FFPE细胞系颗粒的稀释研究,我们确定SNV的LOD为≥5%突变等位基因频率(MAF),Indels的MAF≥10%,SVS的肿瘤纯度≥20%(表1)。duoseq在跑步和运算符精确研究中均达到了> 98%的可重复性(数据未显示)。内部和LAB间的可重复性,并在可比等位基因频率下对事件进行了高度可重复的检测(表2中的数据)。 这些结果表明,杜塞克能够在实验室之间进行均匀的肿瘤分析。内部和LAB间的可重复性,并在可比等位基因频率下对事件进行了高度可重复的检测(表2中的数据)。这些结果表明,杜塞克能够在实验室之间进行均匀的肿瘤分析。

水稻受精卵、成熟胚、未成熟胚再生植株突变的全基因组序列分析
通过全基因组测序,研究了由单个母株的合子、成熟胚和未成熟胚再生的水稻植株 (Oryza sativa L.,‘Nippon-bare’) 的体细胞克隆变异。还对母株和其种子繁殖子代进行了测序。在子代中检测到了 338 个母株序列变异,平均值范围从种子繁殖植株的 9.0 到成熟胚再生体的 37.4。利用种子繁殖植株中的变异计算出的自然突变率为 1.2 × 10 –8,与之前报道的值一致。种子繁殖植株中变异的单核苷酸变异 (SNV) 比例为 91.1%,高于之前报道的 56.1%,且与再生体中的差异不显著。总体而言,如前所述,再生体中 SNV 的转换与颠换比率较低。成熟胚再生的植物的变异明显多于不同子代类型。因此,在水稻遗传操作过程中,使用受精卵和未成熟胚可以减少体细胞克隆变异。

phertilizer:从肿瘤的超低覆盖范围单细胞DNA测序中种植克隆树
新兴的超低覆盖范围单细胞DNA测序(SCDNA-SEQ)技术已经实现了肿瘤内拷贝数畸变(CNA)的高分辨率进化研究。由于这些测序技术非常适合鉴定CNA,由于测序的协调性均匀性,但覆盖范围的稀疏性对研究单核苷酸变体(SNV)的研究构成了挑战。为了最大程度地提高越来越多的超低覆盖范围SCDNA-SEQ数据并获得对肿瘤演化的全面了解,也必须分析SNV从同一组肿瘤细胞中的演变。我们提出了P植物,这是一种从肿瘤的超低覆盖scDNA-seq数据中推断克隆树的方法。基于概率模型,我们的方法通过识别肿瘤史上的关键进化事件来递归对数据进行分区。我们在模拟数据以及两个真实数据集上证明了P生物的性能,发现P Hirtilizer有效地利用了数据中固有的拷贝数信号,以更准确地揭示了与以前的方法相比的克隆结构和基因型。可用性:https://github.com/elkebir-group/phertilizer

水稻受精卵、成熟胚、未成熟胚再生植株突变的全基因组序列分析
通过全基因组测序,研究了由单个母株的合子、成熟胚和未成熟胚再生的水稻植株 (Oryza sativa L.,‘Nippon-bare’) 的体细胞克隆变异。还对母株和种子繁殖子代进行了测序。在子代中检测到了 338 个母株序列变异,平均值范围从种子繁殖植株的 9.0 到成熟胚再生体的 37.4。利用种子繁殖植株中的变异计算出的自然突变率为 1.2 × 10 –8,与之前报道的值一致。种子繁殖植株中变异的单核苷酸变异 (SNV) 比例为 91.1%,高于之前报道的 56.1%,且与再生体中的差异不显著。总体而言,如前所述,再生体中 SNV 的转换与颠换比率较低。成熟胚再生的植物的变异明显多于不同子代类型。因此,在水稻遗传操作过程中,使用受精卵和未成熟胚可以减少体细胞克隆变异。

通过扩展(SBX)/AGBT 2025
sbx =通过扩展进行测序; Q-SCORE = PHRED质量评分,对误差可能性的对数测量; HG001 =一个瓶子(GIAB)财团样本中的基因组,由国家标准技术研究所在多个测序技术中表现出良好的特征; F1分数=变异通话精度的度量,这是精确和回忆的谐波平均值; SNV =单核苷酸变体; indel =插入或删除;双链/单纯形=双链体是指双链DNA的两个链,而单纯形仅指其中一条链。 vcf =变体调用文件

捕获途径 - 口腔学 - 触觉 - 直通...
虽然核分型,鱼类,RT-PCR和微阵列是检测融合基因的常规研究技术,但它们都有局限性。随着基于NGS的方法的改进,DNA和RNA测序迅速成为选择方法。ngs面板促进了同时发现新的变化,以及已知的突变以及基因组研究的结构改变。这种突变检测能力的发展快速增长已超越SNV和Indels,现在包括易位。