XiaoMi-AI文件搜索系统
World File Search Systemfd

摘要。 Nurcahyo FD,Zen HM,Rahma HS,Triyanto A,Yasa A,MD Naim D,Setyawan AD。 2024。局部使用的药用植物的民族植物学研究
摘要。Nurcahyo FD,Zen HM,Rahma HS,Triyanto A,Yasa A,MD Naim D,Setyawan AD。2024。在印度尼西亚中部爪哇省上班加瓦上河上的当地社区使用的药用植物的民族植物学研究。Intl J Bonorowo湿地14:25-36。药用植物都是具有可以治愈某些疾病的特性的植物。世界卫生组织(WHO)指出,世界上有80%以上的人口仍在使用药用植物,包括居住在印度尼西亚班加万河周围的社区。这条河是爪哇河中最长的河流,它在维持生物多样性方面很重要,包括周围的动物群和动植物,尤其是药用植物。这项研究旨在探索上班加旺河(Sidodadi,Ngringo和Palur)沿三个街道中的药用植物的多样性和用途,重点介绍了对当地社区使用的药用植物的文献和理解。90名线人,包括5个密钥和85名普通受访者(21-78岁)。半结构化访谈和参与性观察收集了民族植物学数据,以描述性地介绍并进行定量分析,包括人口统计学,使用价值和线人共识因子。三个街区的村民利用88种药用植物物种进行各种疾病。大多数药用实践都取决于传统方法和口服传播进行知识转移。叶(51.1%)和水果(25%)是使用的主要植物零件,主要通过沸腾(68.5%)和直接消费(23.9%)进行处理。沸腾是将多种植物组合的最优选的方法,因为它被认为是最简单,最具成本效益的方法。Zingiber Officinale Roscoe,Alpinia Galanga(L.)Wild。和Curcuma Longa L.是最常用的植物。这项研究强调了在Bengawan Solo河沿线保存传统的药用植物知识的重要性,以为保护工作,支持社区健康和指导政策,以提供相互利益和生物多样性保护的政策。

硬件 +软件 +工具 +工程
CAN FD Light是基于CAN FD数据链路层的指挥官/响应者通信方法,每个数据框架最多具有64个字节数据字段。它在ISO 11898-1:2024的附件中进行了国际标准化。可以使用FD响应器节点不需要昂贵的外部电路,例如精确的时钟。它们是针对应用程序的,其中一个指挥官节点(正常的CAN CAN协议控制器)管理与多个响应器节点的通信。总线仲裁不是必需的:指挥官节点始终具有通信计划。Bosch的演示者使用了FPGA中实现的公司CAN FD Light IP内核。stmicroelectronics的网络基于其微控制器,其芯片can fd灯光响应者。向量展示了其可以使用的fd灯设计和诊断工具。

骨头
脑衍生的神经营养因子(BDNF)是一种神经营养蛋白,在中枢神经系统和周围组织中表达,受到GSα /CAMP途径的调节。在骨骼中,它调节成骨,并刺激骨化肿瘤(如多发性骨髓瘤)中的RANKL分泌和破骨细胞形成。纤维发育不良(FD)是由GSα基因的功能收益突变引起的罕见的骨骼遗传疾病,其中RANKL依赖性增强的骨吸收是骨骼脆弱性和临床发病率的主要原因。我们观察到BDNF转录本在人类FD病变中表达。具体而言,对从FD患者获得的活检进行的免疫定位研究揭示了成骨细胞中BDNF的表达,并且在纤维组织内的纺锤形细胞中的表现较低。因此,我们假设BDNF可以通过刺激RANKL分泌和骨吸收来在FD的发病机理中发挥作用。为了测试这种疗法,我们使用了人类疾病的EF1α-GSαR201C小鼠模型(FD小鼠)。Western印迹分析显示,与WT小鼠相比,FD小鼠的骨段中BDNF的表达更高,而小鼠FD病变中的免疫标记模式与在人FD中观察到的相似。用抗BDNF的单克隆抗体对FD小鼠进行处理,可减少纤维组织,以及股骨病变内的破骨细胞和骨爆炸的数量。这些结果揭示了BDNF是FD发病机理的新玩家,并且可以在FD骨骼病变中滋养破骨细胞生成的潜在分子机制。他们还建议BDNF抑制作用可能是减少FD中异常骨骼重塑的一种新方法。

硬件 +软件 +工具 +工程
CAN FD Light是基于CAN FD数据链路层的指挥官/响应者通信方法,每个数据框架最多具有64个字节数据字段。它在ISO 11898-1:2024的附件中进行了国际标准化。可以使用FD响应器节点不需要昂贵的外部电路,例如精确的时钟。它们是针对应用程序的,其中一个指挥官节点(正常的CAN CAN协议控制器)管理与多个响应器节点的通信。总线仲裁不是必需的:指挥官节点始终具有通信计划。Bosch的演示者使用了FPGA中实现的公司CAN FD Light IP内核。stmicroelectronics的网络基于其微控制器,其芯片can fd灯光响应者。向量展示了其可以使用的fd灯设计和诊断工具。
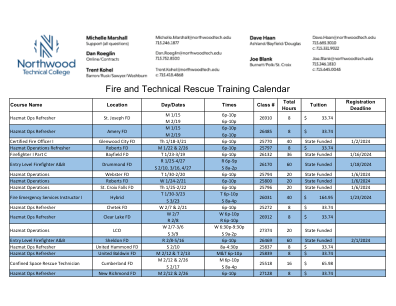
消防和技术救援培训日历
危险品操作复习课 Somerset FD T 2/13 & 2/20 6p-10p 25517 8 33.74 $ 消防员 I 第 C 部分 Rice Lake FD R 2/15-05/02 6:30P-9:30P 27234 36 州政府资助 2/8/2024 SCBA 信心课程 Town of Hayward FD M 2/19 6p-10p 27202 4 19.12 $ 意识复习课 Frederic FD M 2/19 6p-10p 27201 4 19.12 $

针对炎症的载有大麻二酚的纳米胶束可增强口腔黏膜炎的治疗
摘要 口腔黏膜炎(OM)是一种严重的口腔溃疡,是癌症化疗最常见的并发症之一,但其有效治疗仍然是一个严峻的挑战。在本研究中,我们使用脱氧胆酸和岩藻聚糖制备了靶向炎症的纳米胶束(FD),因为岩藻聚糖对P-选择素具有高结合亲和力,可以靶向炎症。然后将疏水性抗炎药物大麻二酚(CBD)负载到FD的疏水核心中。所得的负载CBD的FD胶束(CBD / FD)具有均匀的粒径和形貌,以及良好的血清稳定性。此外,在OM小鼠模型中通过静脉注射或原位滴注给予FD胶束可增强CBD的蓄积和保留。 CBD/FD 在体内局部或全身给药后也表现出比游离 CBD 更好的抗炎作用,同时它们加速 OM 愈合并抑制 Ly6G 炎症细胞浸润和 NF-j B 核转录。我们的结果表明 CBD/FD 纳米胶束是一种很有前途的 OM 治疗剂。

皮肤成纤维细胞表达升高的白介素-8,作为使Fabry病女性疼痛的潜在因素
Fabry病(FD)是X连锁遗传的溶酶体存储障碍。在α-半乳糖苷酶A基因中的突变导致细胞球形甲基甲酰胺(GB3)沉积和两性的触发性疼痛,作为未知病理生理学的早期FD症状。我们旨在阐明皮肤细胞与伤害感受器敏化之间的联系,以性别相关的方式导致FD疼痛。我们使用了27名成人FD患者和20个健康对照组的培养的角质形成细胞和成纤维细胞。培养并进行免疫反应以评估GB3载荷,表皮角质形成细胞和降低的成纤维细胞进行培养和免疫反应。 对疼痛相关的离子通道和促炎性细胞因子的基因表达分析是在降低的成纤维细胞中进行的。 我们进一步研究了诱导的Pluripotent干细胞(IPSC)衍生的具有FD男子的感觉样神经元的电生理特性,并将其健康的男人和米鲁鲁金8(IL-8)或成纤维细胞超级中断作为体外模型Sys-tems孵育。 角质形成细胞没有细胞内,而是膜结合的GB3沉积物。 在很重要的情况下,成纤维细胞显示细胞内GB3,并且与对照组相比,男性和女性在男性和女性中均显示了钾中间/小电导的基因表达较高的基因表达。 此外,细胞因子表达分析显示,仅在雌性FD成纤维细胞中IL-8 RNA水平升高。 斑块夹具研究表明,与IL-8或FD女性的成纤维细胞上清液一起孵育的IPSC神经元细胞系减少了Rheobase Currents。表皮角质形成细胞和降低的成纤维细胞进行培养和免疫反应。对疼痛相关的离子通道和促炎性细胞因子的基因表达分析是在降低的成纤维细胞中进行的。我们进一步研究了诱导的Pluripotent干细胞(IPSC)衍生的具有FD男子的感觉样神经元的电生理特性,并将其健康的男人和米鲁鲁金8(IL-8)或成纤维细胞超级中断作为体外模型Sys-tems孵育。角质形成细胞没有细胞内,而是膜结合的GB3沉积物。在很重要的情况下,成纤维细胞显示细胞内GB3,并且与对照组相比,男性和女性在男性和女性中均显示了钾中间/小电导的基因表达较高的基因表达。此外,细胞因子表达分析显示,仅在雌性FD成纤维细胞中IL-8 RNA水平升高。斑块夹具研究表明,与IL-8或FD女性的成纤维细胞上清液一起孵育的IPSC神经元细胞系减少了Rheobase Currents。我们得出的结论是,女性FD患者皮肤成纤维细胞中的GB3沉积可能导致KCA3.1活性和IL-8分泌增加。这可能导致皮肤伤害感受器的敏化,作为导致性别相关的FD疼痛表型的潜在机制。
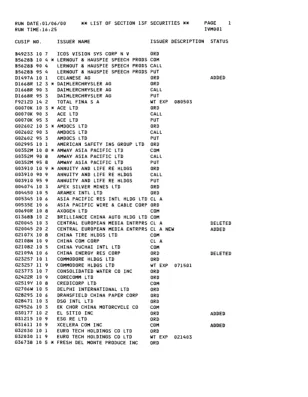
1999 年第 4 季度第 13F 部分证券清单 - SEC.gov
00089C 9 5 7 ADE CORP MASS 0 0 0 9 1 2 1 0 5 ACM GOVT INCOME FD I N C 0 0 0 9 1 4 1 0 1 ACM GOVT SECS FD I N C 000917 1 0 4 ACM GOVT SPECTRUM FD I N C 0 0 0 9 1 8 1 0 2 ACM GOVT OPPORTUNITY FD I N C 000919 1 0 0 ACM MANAGED INCOME FD I N C 000937 1 0 2 ABN AMRO HLDG NV 0 0 0 9 4 2 1 0 2 ACM MUN SECS INCOME FD I N C 0 0 0 9 4 5 AC 9 ADT OPERATIONS 000949 1 0 7 ACM 管理的 DLR 收入 FD 000950 1 0 5 * AFC 有线系统 I N C 000950 9 0 5 AFC 有线系统 I N C 000950 9 5 5 AFC 有线系统 I N C 0 0 0 9 5 5 1 0 4 ACT TELECONFERENCING I N C 000957 1 0 0 * ABM INDS I N C 000957 9 0 0 ABM INDS I N C 000957 9 5 0 ABM INDS I N C 0 0 0 9 7 3 1 0 7 ACT MFG I N C 0 0 0 9 7 5 1 0 2 * ACT NETWORKS I N C 0 0 0 9 7 5 9 0 2 ACT NETWORKS I N C 0 0 0 9 7 5 9 5 2 ACT NETWORKS I N C 0 0 1 0 3 1 1 0 3 AEP INDS I N C 0 0 1 0 5 5 1 0 2 * AFLAC I N C 0 0 1 0 5 5 9 0 2 AFLAC I N C 0 0 1 0 5 5 9 5 2 AFLAC I N C 0 0 1 0 8 4 1 0 2 * 爱科集团 0 0 1 0 8 4 9 0 2 爱科集团 001084 9 5 2 爱科集团 0 0 1 2 0 4 1 0 6 * AGL RES I N C 001204 9 0 6 AGL RES I N C 001204 9 5 6 AGL RES I N C 0 0 1 2 5 0 1 0 9 AG SVCS AMER I N C 0 0 1 2 9 6 1 0 2 AHL SVCS I N C
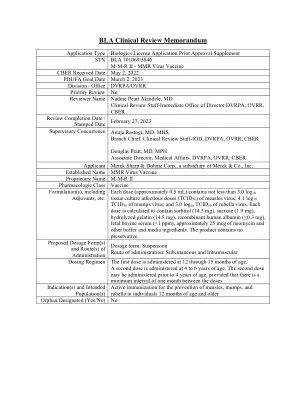
麻疹,腮腺炎和风疹病毒疫苗活
ACIP免疫实践咨询委员会AE不利事件AM AM AM BLA BLA生物制品CBER生物学评估和研究中心CCID 50细胞培养感染剂量50%CDISC临床数据互换标准CI CI置信度CI置信区CI间隔ELISA酶ELISA酶ELISA酶的IMESA酶良好的欧洲工会良好的欧洲工会EU EU EUSIT ASSITIAL EUSITIAL EU UNION ASSAY EU FOF FEF FACS ASS ASS ASS ASS ASS ASS ASS APC ASS APS APS IS FD FD ASS APS IS与FD UD FD UD FD AS SET FD ASS SET FD ASS ST SOT FD ASS ST STE几何平均滴度GMTR几何均值比率GPELISA糖蛋白ELISA IGG免疫球蛋白G IM肌肉内IND IND研究新药物IU国际ll下限MIU MIU MILI MILI-INTERMATION forming units PPS Per Protocol Set PPS1 Per Protocol Set Post-dose 1 PPS2 Per Protocol Set Post-dose 2 PREA Pediatric Research Equity Act rHA recombinant human albumin RoA route of administration SAE serious adverse event SC subcutaneous SmPC Summary of Product Characteristics SRR seroresponse rate TCID 50 tissue culture infectious dose 50% USPI US Prescribing Information WFI water for injection

2021-22 年度报告 - Shakti Pumps
eq> s; g crkrs gq,cm+h [kq'gk jgh gs fd bl qk; usaf; usaf; y bzvj 2021&22 esa vkids“ kfdr ieil〜1⁄4bf.m; k1⁄2 unkj jgk gs] ekjk fxzm&dusdvsm lksyj iei esa gs ftlls ikuh vksj mtkz nksukas nksukas dh cpr gksxha fmekam dks /;ku esa j[krs gq, gekjh daiuh us tks dh gekjs R&D Vhe us xzkgd dh mPp fjDOk;kjesUV dks ns[krs gq,] ikuh vkSj fctyh dh cpr dks ns[msagrs gqV,] Vhe Ir gqvk gSA S4RM VsDuksykWth esa geus cgqr vPNk dk;Z fd;k gS exVj ,fQf”k,alh dks bEizwo fd;k gSA gekjs xzkgdksa dk fctyh dk fcy de djuk geksjk FkxVh Vykhe] kkunkj dk;Z fd;k vkSj nwljs isVsaV esa ,uthZ ,fQf”k,a”kh dks ,pho fd;kA gekjs lkeus ,d pSysat fctyh daiuh ds ykWlsl dk Fkk] geuft gezwos jzvos dgsjV Q]g pk one dk;Z fd;k vkSj cgqr vPNh VsDuksykWth Msoyi gqbZ vkSj gesa isVsaV izkIr gqvkA