XiaoMi-AI文件搜索系统
World File Search Systempipeline

辉瑞管道
实际结果可能与此类陈述明示或暗示的结果存在重大差异。对于管道产品,无法保证临床研究会成功,产品会进入下一开发阶段,产品会获得必要的监管批准或证明其在商业上取得成功。如果基本假设被证明不准确或风险或不确定性成为现实,实际结果可能与前瞻性陈述中规定或暗示的结果存在重大差异。有关这些因素和其他因素的更多信息,可在辉瑞截至 2023 年 12 月 31 日财年的 10-K 表年度报告及其后续 10-Q 表报告中找到,包括其中标题为“风险因素”和“前瞻性信息和可能影响未来结果的因素”的部分,以及辉瑞后续的 8-K 表报告,所有这些报告均已提交给美国证券交易委员会,可在 www.sec.gov 和 www.pfizer.com 上查阅。

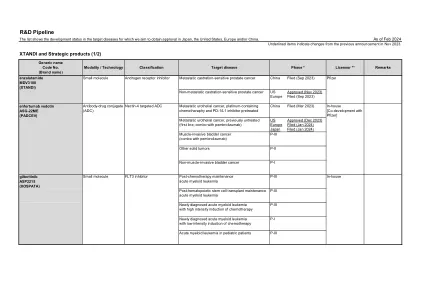
Pfizer Pipeline
●本演示文稿包括具有重大风险和不确定性的前瞻性陈述,这些陈述可能导致实际结果与此类陈述所表达或暗示的结果有实质性差异。不能保证临床研究成功的管道产品,这些产品将获得必要的监管批准,或者证明它们将在商业上取得成功。如果基本假设证明不准确,风险或不确定性实现,则实际结果可能与前瞻性陈述所规定的或暗示的结果有重大差异。Additional information regarding these and other factors can be found in Pfizer's Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in our subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov和www.pfizer.com。
Astellas Pharma Inc.
以前公告(2023年11月)的更新:恩扎拉胺:2023年11月在美国批准了非转移性cast割敏感的前列腺癌,并具有生化复发,并以高风险转移。Enfortumab Vedotin:2023年12月在美国批准,并于2024年1月在欧洲和日本提交,以在一线环境中为当地晚期或转移性尿路上皮癌。fezolitant:2023年12月在欧洲批准了与更年期相关的中度至重度血管舒张症症状。在日本进入第3阶段的血管舒适症状与更年期有关。avacincaptad pegol:2023年8月,删除了美国对与年龄相关的黄斑变性的地理萎缩的描述。

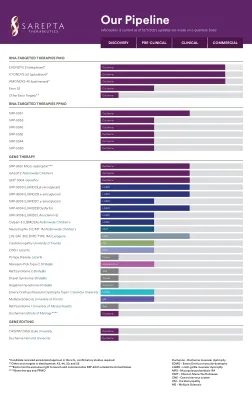
Pfizer Pipeline
●本演示文稿包括具有重大风险和不确定性的前瞻性陈述,这些陈述可能导致实际结果与此类陈述所表达或暗示的结果有实质性差异。不能保证临床研究成功的管道产品,这些产品将获得必要的监管批准,或者证明它们将在商业上取得成功。如果基本假设证明不准确,风险或不确定性实现,则实际结果可能与前瞻性陈述所规定的或暗示的结果有重大差异。Additional information regarding these and other factors can be found in Pfizer's Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in our subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov和www.pfizer.com。
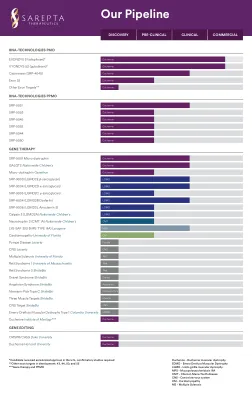

辉瑞管道
实际结果可能与此类陈述明示或暗示的结果存在重大差异。对于管道产品,无法保证临床研究会成功,产品会获得必要的监管批准或被证明具有商业成功。如果基本假设被证明不准确或风险或不确定性成为现实,实际结果可能与前瞻性陈述中规定或暗示的结果存在重大差异。有关这些和其他因素的更多信息,可在辉瑞截至 2023 年 12 月 31 日财年的 10-K 表年度报告及其后续 10-Q 表报告中找到,包括其中标题为“风险因素”和“前瞻性信息和可能影响未来结果的因素”的部分,以及我们后续的 8-K 表报告,所有这些报告均已提交给美国证券交易委员会,可在 www.sec.gov 和 www.pfizer.com 上查阅。

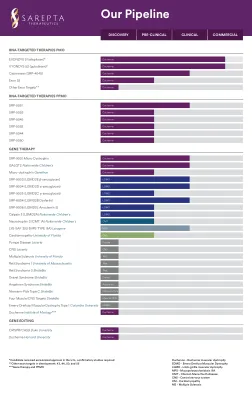
辉瑞管道
● 本报告包含前瞻性陈述,这些陈述受重大风险和不确定性的影响,可能导致实际结果与这些陈述所表达或暗示的结果大不相同。对于管道产品,无法保证临床研究会成功,产品会获得必要的监管批准,或产品会证明具有商业成功。如果基本假设被证明不准确或风险或不确定性成为现实,实际结果可能与前瞻性陈述中所述或暗示的结果大不相同。有关这些因素和其他因素的更多信息,可在辉瑞截至 2022 年 12 月 31 日的财政年度的 10-K 表年度报告及其后续的 10-Q 表报告中找到,包括其中标题为“风险因素”和“前瞻性信息和可能影响未来结果的因素”的章节,以及我们后续的 8-K 表报告,所有这些报告均已提交给美国证券交易委员会,可在 www.sec.gov 和 www.pfizer.com 上查阅。

BCPUD管道
有兴趣了解有关BCPUD的更多信息吗?参加每月一次的董事会会议,观看会议视频或阅读会议记录 - 在线和面对面。会议:BCPUD在每个月的第三个星期三举行常规董事会会议。邀请公众参加面对面或Zoom参加会议。会议议程在董事会会议之前的每个星期五将上传到我们网站的议程页面。除了议程外,公众还可以访问与议程项目相关的董事会会议材料。材料通常是在会议前1-2天加载的。请访问www.bcpud.org,然后单击董事会会议议程链接以访问该页面。视频:每次会议后,BCPUD将每月的董事会会议缩放视频上传到我们的网站。可以在我们网站的会议记录页面上找到vid-eos。也可以在我们的网站上找到指向董事会会议记录页面的链接。会议分钟:BCPUD在最终确定会议记录后(通常在下一个董事会会议上)将每个董事会会议的会议记录的副本上传到我们的网站。简而言之,公众可以通过多种方式观看和/或查看每月董事会会议。请花点时间查看我们的网站,并了解有关BCPUD正在发生的事情的更多信息。