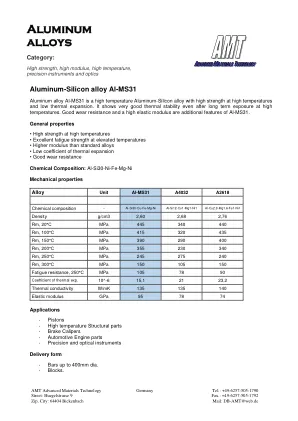
XiaoMi-AI文件搜索系统
World File Search System模量


用CFRP
世界各地的钢结构都容易在其使用寿命期内恶化。这种恶化可以分别由疲劳负荷和极端天气条件引起的裂缝和腐蚀引起的钢构件的潜在强度和刚度。此外,在设计和施工阶段可能会出现缺陷。进行钢结构改造的常规方法是使用焊缝或螺栓连接到结构的钢板[1]。但是,这种方法呈现出缺点,包括焊接施加的残余应力,这可能会对结构造成新的损害[2,3]。此外,钢板容易受到腐蚀的影响,其重量重量在安装过程中构成了挑战[4,5]。另外,将外部粘结碳纤维增强聚合物(CFRP)的应用可以提供耐用的解决方案来应对这些挑战[6,7]。CFRP材料的高强度重量比和耐腐蚀性在选择改造钢组件的选择中具有重要作用[8-10]。近年来,高级复合材料的应用在改造民用基础设施方面已获得接受。在这些类型的材料中,CFRP和石墨纤维增强聚合物(GFRP)已得到很好的确定[11]。但是,由于其强度较高,CFRP表现出优于GFRP的优势。研究表明,CFRP改造系统可以有效地增强钢构件的弯曲能力并延长其疲劳寿命[4,12 - 32]。CFRP根据其弹性模量分类为低模量(LM),正常模量(NM)或中间模量(IM),高模量(HM)和超高模量(UHM)。没有一种一致的方法来表征每个类别的弹性模量范围。但是,它可以相对于表1所示的钢弹性模量表示[33]。

光学,电气和机械性能
生物聚合物是有前途的材料,如果其低机械和生物活性特性都得到改善,则可以在骨骼替代应用中广泛使用。在这方面,这项研究的主要目的是改善机械和生物学特性,除了改善光学和电气特性以适合于裂缝愈合目的使用。因此,在这项研究中,将一批聚(乙烯基醇; PVA)和生物学提取的羟基磷灰石(BHA)机械地以(70:30 vol。%)为准。然后,将氧化镁(MGO)和碳化硅(SIC)添加到该批次中,其体积百分比不同,在120°C时加热。测量了物理,机械,光学和电气性能。此外,通过将它们浸入模拟的体液(SBF)中,然后通过扫描电子显微镜(SEM)进行检查,从而评估了这些样品在其表面上形成磷灰石层的能力。获得的结果澄清说,由于这些添加剂的添加剂,改善了微度,压缩强度,Young的模量,纵向模量,纵向模量,大量模量和剪切模量的机械性能。也观察到,BHA和MGO纳米颗粒的存在增强了准备样品的生物活性,光学和电性能。获得的结果令人鼓舞,这项研究的目的已成功实现。




m在使用扭矩电效应
多模式触觉感知对于增强现实应用中的感知体验至关重要。迄今为止,已经开发出几个人造触觉接口来感知压力和前接触信号,同时以量化的模量检测物体类型和柔软度仍然具有挑战性。在这里,受昆虫触角上的坎帕形感觉的启发,我们提出了一种半球双峰智能触觉传感器(位)阵列,该阵列使用了落压效果。该系统能够识别柔软度,模量定量和材料类型识别。原则上,由于材料的变形性变化,与测试对象接触时,钻头生成的零件唯一的摩擦式输出指纹。此外,由于不同的电子亲和力,钻头阵列可以准确识别材料类型(精度为99.4%),促进柔软度识别(100%精度)和模量定量。有望基于摩擦效应的位具有小型化的潜力,以提供实时准确的触觉信息,作为人工天线,用于人工机器整合的应用。

低温聚合物复合材料研究进展...
因此,本研究首先简要介绍一些重要的机械测试方法,然后概述聚合物复合材料,最后描述聚合物复合材料在暴露于低温时强度、模量、韧性、脆性和热导率等性能的变化,并与强度、模量、韧性、脆性和热导率等类似性能进行比较。还重点介绍了聚合物复合材料的机械和热性能的不同表征数据,以评估其是否适合低温应用,这将作为一份关于温度变化对低温范围内改性聚合物性能影响的综合报告,使人们熟悉聚合物复合材料在低温下的性能和行为。

定时侧通道通过自动化程序维修
1 modpow(基本,指数,模量,宽度){2 biginteger r0,r1 = biginteger.one,base; 3 for(int i = 0; i