XiaoMi-AI文件搜索系统
World File Search System溴化乙锭

焊接添加剂制造:文献审查22β-羟基丁酮通过抑制MMP -9活性和MAPK信号传导
Ethnopharmacological relevance: 22 β -hydroxytingenone (22-HTG) is a quinonemethide triterpene isolated from Salacia impressifolia (Miers) A. C. Smith (family Celastraceae), which has been used in traditional medicine to treat a variety of diseases, including dengue, renal infections, rheumatism and cancer.但是,尚未阐明22-HTG和黑色素瘤细胞中潜在的分子机制的抗癌作用。研究的目的:本研究研究了SK-MEL-28人黑色素瘤细胞中22-HTG的凋亡诱导和抗转移性势力。材料和方法:首先,评估了22-HTG在培养的癌细胞中的体外细胞毒性活性。然后,使用黑色素瘤细胞(SK-MEL-28)中的锥虫蓝色测定法确定细胞活力,其随后是细胞周期,膜联蛋白V-FITC/碘化碘化物测定(膜联蛋白/PI),以及用于评估线粒体膜潜力的测定,使用反应性牛(Recatige oxygen cytomry)评估线粒体膜潜力。使用Acridine橙/溴化乙锭(AO/BE)染色的荧光微副本。RT-QPCR以评估BRAF,NRA和KRAS基因的表达。在重建的人类皮肤的三维(3D)模型中评估了22-HTG的抗侵入性潜力。结果:22-HTG降低了SK-MEL-28细胞的生存力,并导致形态学变化,因为细胞收缩,染色质凝结和核碎裂。此外,22-HTG引起了凋亡,通过用AO/BE和Annexin/pi染色来证明,这证明了这一点。凋亡可能是由于线粒体不稳定性引起的,而无需ROS产生。通过22-HTG处理降低了BRAF,NRA和KRAS的表达,它们是黑色素瘤发育中重要的生物标志物。在重建的皮肤模型中,22-HTG能够降低真皮中黑色素瘤细胞的侵袭能力。

分子生物学中的技术 - 质粒DNA的方法...
t eChniquers i n M Olecular b Iology - 用于P LASMID DNA I求解DNA分离的方法:分子生物学技术在复杂基因组分析中的应用取决于准备纯质粒DNA的能力。大多数质粒DNA隔离技术有两种口味,简单 - 低质量的DNA制剂,更复杂,耗时但高质量的DNA制剂。对于许多DNA操作,例如限制酶分析,亚克隆和琼脂糖凝胶电泳,简单的方法就足够了。大多数DNA测序,PCR操作,转换和其他技术都需要高质量的制剂。大多数方法都以大量细菌细胞开头,这些细菌细胞包含选择的质粒并离心至颗粒。然后,细胞在基本条件下通过洗涤剂钠硫酸盐(SDS)的混合物裂解,或添加蛋白酶(溶菌酶)以削弱和破坏宿主细胞壁。这两种方法的结果都导致紧凑型超螺旋质粒DNA分子释放到溶液中。下一个问题是将RNA,基因组DNA和其他细胞成分与细胞分开。如何完成此操作取决于所使用的方法。碱性裂解制剂是隔离少量质粒DNA的最常用方法,通常称为小型质子。此方法将SDS用作弱洗涤剂,以在NaOH存在的情况下使细胞变性,该清洁剂可将细胞壁和其他细胞分子水解起来。高pH值通过添加乙酸钾进行中和。这将质粒DNA和RNA留在溶液中。钾对样品有额外的影响。钾离子与SD相互作用,使其成为不溶性的洗涤剂。SD会很容易沉淀,并且可以通过离心分离。这样做的不溶性SDS会捕获较大的基因组DNA并将其从上清液中清除。通常通过添加RNASEA消化去除RNA。这仅留下溶液中的蛋白质,碳水化合物和RNA核苷单体。原发性醇(例如乙醇或丙醇)用于沉淀DNA。这是通过对水的重新排序来实现的,使DNA聚集体并变得不溶性。结果是一种纯净的DNA颗粒,可以重悬于温和缓冲的溶液或水中。建议使用大量培养物中煮沸的微型REIPREP来制备少量的质粒DNA。虽然此方法非常快,但产生的DNA质量低于碱性裂解小型培训的质量。在碱性裂解小型方法中,溶菌酶用于水解负责使细菌细胞壁具有其强度的广泛交联蛋白。然后将细胞煮沸以进一步使蛋白质结染并破坏细胞壁。然后用酒精沉淀质粒DNA。这两种方法都将仅产生几µg质粒DNA。对于纯度较高的较大数量,需要许多其他步骤。通过在非常高的重力力下在氯化丘密度梯度中离心,根据其密度分离其密度。氯化剖腹梯度产生的高质量质粒DNA不含大多数污染物,但使用溴化乙锭来识别DNA(潜在的诱变剂),并且需要长时间的超级离心运行以建立密度梯度。该方法是通过使用碱性裂解方法裂解细胞的,并在350,000 x g下离心14小时。首先,将CSCL梯度在小管中制成,并用溴化乙锭添加DNA。在旋转时,DNA将向下迁移,直到达到与质粒相同的CSCL的密度。因此,较大的DNA将与紧凑的质粒DNA分离。用紫外线可视化质粒带,用针切除,然后重复该过程。您可以看到,这是一种非常复杂且乏味的方法,用于隔离DNA,通常不经常在柱分离的出现中使用。现在存在一种更流行的方法,它利用了质粒DNA的物理特性和碱性裂解方法中发现的污染物的差异。核酸是负电荷的,因此可以使用阴离子交换

补充图1
补充图 1 | ERD7 转基因拟南芥突变株系的生成和表征。(A)根据 TAIR 提供的信息,描绘了拟南芥 ERD7 ORF (AT2G17840.1) 的插图,左侧为 5' 端。标示了 ERD7 基因外显子(框)和内含子(线)以及在相应的单拷贝、T 3 纯合 erd7-1 和 erd7-2 突变株系中针对 CRISPR/Cas9 基因组编辑的 T-DNA 插入和 sgRNA 区域的相对位置。还显示了 (C) 中用于基因分型和 RT-PCR 分析的引物对的相对位置。 (B) 野生型和 erd7-2 突变株系中 ERD7 基因和蛋白质的核苷酸和推导多肽序列的比较,表明 ERD7 基因(和相应的转录本)中预期有 1711 个核苷酸缺失,导致 erd7-2 突变株系中编码蛋白质有 398 个氨基酸缺失。ERD7 野生型和 erd7-2 突变体 DNA 序列中 CRISPR/Cas9 原间隔区相邻基序带下划线。使用 ClustalO 算法 (ebi.ac.uk/Tools/msa/clustalo) (Madeira et al., 2019) 对野生型和突变型 ERD7 蛋白质的推导氨基酸序列进行比对。(C) erd7-1 和 erd7-2 突变株系的 PCR 和 RT-PCR 分析。图中显示的是从 15 天大的 WT、erd7-1 和 erd7-2 植物的莲座叶中提取的 gDNA 的 PCR 分析(上图)和 mRNA 的 RT-PCR 分析(下图),并使用所示引物对进行评估;引物对的位置参见 (A)。有关用于生成和表征两个 erd7 突变系的所有引物序列,另请参阅补充表 1。通过 DNA 凝胶电泳和溴化乙锭染色分析 PCR 产物和 RT-PCR 产物。注意,在两个突变系中均不存在分别对应于 ERD7 基因和 ERD7 表达(即转录本)的 PCR 和 RT-PCR 产物,而 erd7-1 突变体中存在 T-DNA,这与预期一致。拟南芥 TUB4 用作内源对照。
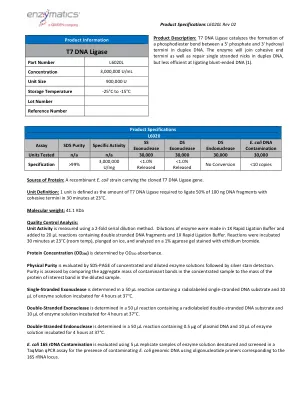
T7 DNA连接酶
蛋白质的来源:一种带有克隆的T7 DNA连接酶基因的重组大肠杆菌菌株。单位定义:1个单位定义为在30分钟内在23°C下在30分钟内将100 ng DNA片段的50%结合的T7 DNA连接酶的量。分子量:41.1 kDa质量控制分析:使用2倍连续稀释方法测量单位活动。稀释液,并添加到含有双链DNA片段和1倍快速连接缓冲液的20 µL反应中。在23°C(室温)下孵育30分钟,浸在冰上,并在用溴化乙锭染色的1%琼脂糖凝胶上进行分析。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。大肠杆菌16S rDNA的污染是使用5 µL r菌酸溶液的样品变性的样品,并在Taqman QPCR分析中筛选,以使用与16S rRNA locus相应的寡核苷酸引物,使用污染的大肠杆菌Genomic DNA。

什么是DNA隔离定义
DNA提取自1869年弗里德里希·米舍(Friedrich Miescher)首次试图隔离它以来,它已经走了很长一段路,意外地发明了一种核酸隔离方法,后来其他人会完善。该过程涉及分解细胞膜和核信封以获得DNA,这对于PCR,DNA克隆,测序和电泳等各种分子生物学应用至关重要。取决于样本类型 - 需要植物,血液,细菌或其他 - 需要不同的提取方法,每种方法都有自己的手术,以确保DNA的所需纯度和数量。从苯酚 - 氯仿等醇到蛋白酶K,CTAB,自旋柱,磁珠等等,存在各种技术,每个技术都基于样品的特定要求选择。DNA提取的故事是连续的创新和精致之一,像Meselson和Stahl这样的先驱在1958年建立了全功能程序,而其他人则随着时间的推移贡献了他们的方法。从细胞中提取DNA的过程涉及三个主要步骤:细胞裂解,沉淀和溶解DNA。所使用的化学或组合的类型可能会根据目标和细胞类型而有所不同。对于柔软的细胞壁,如在结核分枝杆菌中发现的,用简单的裂解缓冲液加热是有效的。然而,较硬的细胞壁需要机械,化学和酶促方法来提取DNA。基于化学的DNA提取方法是基于溶液的,涉及各种有机和无机溶液。这些包括SDS,CTAB,苯酚,氯仿和硫氰酸鸟酯。所有程序的主要步骤是细胞裂解,降水和洗脱。基于溶液的(化学)DNA提取进一步分为基于有机溶剂的和基于无机溶剂的方法。有机溶剂(如苯酚和氯仿)以前已被使用,但由于危险而灰心。相反,采用了更安全的替代方案,例如Triton X100和EDTA。不同的化学物质有特定目的;蛋白酶K等酶分解蛋白质直接靶向氨基酸连接。DNA提取过程的有效性可能受细胞类型的影响以及某些化合物或化学物质组合的使用。在冷链中,尽管有一些缺点,但基于蛋白酶K的DNA分离方法还是有效的方法。此过程的一个问题是酶的稳定性降低,随着时间的流逝而降低。使用无机溶液(例如氯化钠和乙酸钾)与蛋白酶K结合使用的盐溶液。但是,提取的DNA的纯度可能是一个问题,因为尽管获得了足够的质量,但收益率可能不会令人满意。苯酚 - 氯仿 - 异氧化酒精法(PCI)是提取DNA的另一种流行技术。它使用液化缓冲液,苯酚和氯仿对蛋白质和破坏细胞,从而产生出色的产率和纯度。可以通过使用现成的DNA提取缓冲区来修改此方法,从而快速而简单。高质量的DNA产量和简单操作系统对于准确的DNA分析至关重要。相反,基于二氧化硅的DNA提取方法提供了一种独特的方法,依赖于二氧化硅和DNA相互作用的独特化学。带正电荷的二氧化硅颗粒在离心过程中与带负电荷的DNA结合,从而允许高质量的DNA产量和易于操作。由于其简单性和有效性,该市售技术已在诊断实验室中被广泛接受。为了验证提取的DNA,可以使用溴化乙锭或其他与DNA在UV光下反应的荧光染料在琼脂糖凝胶上电泳。通过计算260 nm和280 nm波长的吸光度来测量DNA的纯度。确定DNA纯度的最常见方法是A260/A280比率,对于优质DNA,应在1.7-2.0之间。较低的比率表明存在更多的污染物。荧光测量是确定DNA产量和浓度的另一种流行方法,它由于其广泛的可用性和比吸光度方法更高的灵敏度。也可以使用二苯胺(DPA)指示器确认DNA的存在,该指标涉及DNA化学水解。与分光光度计在600 nm处的吸光度强度也可以通过将DNA浓度与已知DNA浓度的标准曲线进行比较来确定DNA浓度。已开发和应用多种用于DNA提取的方法,包括用于传染病的护理核酸测试和人类DNA提取方法。方法的选择取决于DNA的特定应用和所需的质量。