XiaoMi-AI文件搜索系统
World File Search System磷酸化

menin 抑制剂 ziftomenib 与伊马替尼产生协同作用......
图 4. 齐托美尼和伊马替尼联合治疗消除了 KIT 蛋白表达,关闭了 AKT/mTOR 和 ERK 通路,并强烈诱导了细胞周期停滞和凋亡。A. GS11331 模型中的药效学 (PD) 研究的肿瘤生长曲线。用载体 (QD)、齐托美尼 (1X,QD)、伊马替尼 (100 mpk,QD) 和联合用药治疗肿瘤。在第 5 天和第 8 天收获肿瘤。B. 总或磷酸化 KIT 蛋白的免疫印迹,指示 MAPK/PI3K 通路成分和凋亡标志物。联合治疗导致总 KIT 蛋白消失,AKT 活性/靶标磷酸化 (磷酸化的 mTOR 和 AKT) 受到强烈抑制,细胞周期停滞 (RB 磷酸化) 和细胞死亡 (裂解 PARP)。与第 5 天相比,第 8 天对联合治疗的信号反应更加深化。

相关激酶TBK1:评估NCOA7
图5。TBK1和IKKβ结构域组织的结构比较。 a)TBK1 KU D135N结构,显示了激酶结构域(KD)和泛素样域(ULD),具有插图的TBK1和IKKβKD。 (b)位于IKKβ杂质内的TBK1 KU D135N结构显示与IKKβ的支架二聚域(SDD)的兼容性。 与SDD相互作用的残基以紫色突出显示。 注意。 改编自“转载体磷酸化的储罐结合激酶1的分子基础”。 https://doi.org/10.1073/pnas.1121552109。 Ser172的磷酸化触发TBK1激活所需的构象变化,TBK1和IKKβ结构域组织的结构比较。a)TBK1 KU D135N结构,显示了激酶结构域(KD)和泛素样域(ULD),具有插图的TBK1和IKKβKD。(b)位于IKKβ杂质内的TBK1 KU D135N结构显示与IKKβ的支架二聚域(SDD)的兼容性。与SDD相互作用的残基以紫色突出显示。注意。改编自“转载体磷酸化的储罐结合激酶1的分子基础”。https://doi.org/10.1073/pnas.1121552109。 Ser172的磷酸化触发TBK1激活所需的构象变化,https://doi.org/10.1073/pnas.1121552109。Ser172的磷酸化触发TBK1激活所需的构象变化,Ser172的磷酸化触发TBK1激活所需的构象变化,

氧化还原生物学
饮食原腺苷(PAC)消费量与结直肠癌(CRC)的风险降低有关。在包括CRC在内的人类癌症中,表皮生长因子(EGF)受体(EGFR)信号通路的失调频率很高。我们先前表明六聚体PAC(十六进制)在人CRC细胞中发挥抗增殖和凋亡作用。这项工作是否可以通过调节EGFR途径的能力发挥抗CRC效应。在增殖的CACO-2细胞中,十六进制的作用抑制了EGF诱导的EGFR二聚化和NADPH氧化酶依赖性磷酸化,在Tyr 1068时磷酸化,降低了EGFR在脂质筏上的位置,并抑制了促促进性和抗磷酸和抗蛋白质路线的下部激活RAF/MEK/ERK1/2和PI3K/AKT。在不存在和存在EGF的情况下, HEX还促进了EGFR的内在化。 虽然HEX在Tyr 1068时降低了EGFR磷酸化,但EGFR Tyr 1045磷酸化增加了。 后者为泛素连接酶C-CBL提供了一个对接位点,并通过溶酶体促进EGFR降解。 重要的是,HEX与靶向EGFR的化学治疗药物Erlotinib协同作用,均以降低EGFR磷酸化和抑制细胞生长的能力。 因此,饮食PAC可以通过通过氧化还原和非雷多斯调节的机制调节EGFR促进性信号通路来发挥抗CRC作用。 此外,十六进制还可以增强以EGFR为目标的药物的作用。HEX还促进了EGFR的内在化。虽然HEX在Tyr 1068时降低了EGFR磷酸化,但EGFR Tyr 1045磷酸化增加了。后者为泛素连接酶C-CBL提供了一个对接位点,并通过溶酶体促进EGFR降解。重要的是,HEX与靶向EGFR的化学治疗药物Erlotinib协同作用,均以降低EGFR磷酸化和抑制细胞生长的能力。因此,饮食PAC可以通过通过氧化还原和非雷多斯调节的机制调节EGFR促进性信号通路来发挥抗CRC作用。此外,十六进制还可以增强以EGFR为目标的药物的作用。

RPT6的磷酸化控制其结合DNA并调节在记忆形成期间雄性大鼠海马中基因表达的能力
记忆形成需要协调控制基因表达,蛋白质合成和泛素 - 蛋白酶体系统(UPS)介导的蛋白质降解。UPS的催化成分,26S蛋白酶体包含由两个19S调节帽的20S催化核心,以及在丝网上120(PRPT6-S120)的19S CAP调节子基RPT6的磷酸化已广泛与控制活性依赖性依赖性依赖性蛋白酶体活动有关。最近,还显示RPT6在记忆形成期间在海马中具有类似转录因子的作用的蛋白酶体外作用。然而,对于大脑中“ Free” RPT6的蛋白酶体无关函数,在记忆形成期间以及该转录控制功能是否需要S120的磷酸化。在这里,我们使用了RNA测序以及新型的遗传方法以及生化,分子和行为测定方法来检验以下假设:PRPT6-S120在内存形成过程中prpt6-S120的独立性独立于蛋白酶体来结合DNA并调节基因表达。rNA介导的siRNA介导的自由RPT6敲低后的序列显示,在恐惧状态下,男性大鼠的背侧海马中有46个基因靶标,其中RPT6参与转录激活和抑制。通过RISPR-DCAS9介导的RPT6在靶基因上的人工放置,我们发现单独的RPT6 DNA结合对于改变学习后改变基因表达可能很重要。此外,CRISPR-DCAS13介导的S120转化为RPT6上的甘氨酸表明,S120处的磷酸化是RPT6结合DNA并在记忆形成过程中正确调控转录的必要条件。一起,我们揭示了RPT6在控制记忆形成过程中控制基因转录中磷酸化的新功能。

TIFAB通过形成与TIFA
核因子κB(NF -κB)被各种炎症和传染性分子激活,并参与免疫反应。已经阐明了ADP-β-D-甘露糖(ADP- HEP),一种革兰氏 - 阴性细菌的代谢物,通过α-激酶1(ALPK1) - TIFA -TIFA -TRAF6信号传导激活NF -κB。ADP- HEP刺激ALPK1的激酶活性用于TIFA磷酸化。 磷酸化 - 依赖性TIFA低聚物和TRAF6之间的复合形成促进了TRAF6对NF -κB激活的多泛素化。 tifab是缺乏磷酸化位点和TRAF6结合基序的TIFA同源物,是TIFA -TRAF6信号传导的负调节剂,与髓样疾病有关。 TIFAB被指出通过与TIFA和TRAF6的相互作用来调节TIFA -TRAF6信号传导。但是,对其生物学功能知之甚少。 我们认为TIFAB与TIFA二聚体形成复合物,TIFA二聚体是NF -κB激活涉及的TIFA的固有形式,而是与单体TIFA。 TIFA/TIFAB复合物以及基于生化和细胞的分析的结构分析表明,TIFAB形成具有TIFA的稳定异二聚体,抑制TIFA二聚体的形成,并抑制TIFA – TRAFAFAF6信号传导。 所得的TIFA/TIFAB复合物是缺少磷酸化位点的“伪-TIFA二聚体”,在TIFAB中缺乏TRAF6结合基序,无法形成针对NF -K -κB活化涉及的磷酸化TIFA寡聚的有序结构。 这项研究阐明了TIFAB通过TIFA-TRAF6信号进行调节的分子和结构基础。ADP- HEP刺激ALPK1的激酶活性用于TIFA磷酸化。磷酸化 - 依赖性TIFA低聚物和TRAF6之间的复合形成促进了TRAF6对NF -κB激活的多泛素化。tifab是缺乏磷酸化位点和TRAF6结合基序的TIFA同源物,是TIFA -TRAF6信号传导的负调节剂,与髓样疾病有关。TIFAB被指出通过与TIFA和TRAF6的相互作用来调节TIFA -TRAF6信号传导。但是,对其生物学功能知之甚少。我们认为TIFAB与TIFA二聚体形成复合物,TIFA二聚体是NF -κB激活涉及的TIFA的固有形式,而是与单体TIFA。TIFA/TIFAB复合物以及基于生化和细胞的分析的结构分析表明,TIFAB形成具有TIFA的稳定异二聚体,抑制TIFA二聚体的形成,并抑制TIFA – TRAFAFAF6信号传导。所得的TIFA/TIFAB复合物是缺少磷酸化位点的“伪-TIFA二聚体”,在TIFAB中缺乏TRAF6结合基序,无法形成针对NF -K -κB活化涉及的磷酸化TIFA寡聚的有序结构。这项研究阐明了TIFAB通过TIFA-TRAF6信号进行调节的分子和结构基础。

胃生素可以改善N2A/APP细胞中的突触障碍,线粒体功能障碍和氧化应激
阿尔茨海默氏病的特征是异常的β-淀粉样蛋白和TAU积累,线粒体功能障碍,氧化应激和突触功能障碍。在这里,我们旨在评估胃生育(一种酚类糖苷)对表达人类瑞典突变APP(N2A/APP)的鼠神经母细胞瘤N2A细胞的胃生育作用的机制和信号传导途径。我们发现胃生蛋白增加了突触前SNAP,突触pos蛋白和突触后-PSD95的水平,并降低了N2A/APP细胞中APP和β1-42水平的磷酸化磷酸化Ser396和β1-42水平。胃生素治疗减少了活性氧的产生,脂质过氧化,线粒体碎片和DNA氧化;恢复的线粒体膜电位和细胞内ATP产生。上调的磷酸化 - GSK-3β以及磷酸化和磷酸-JNK的降低参与胃生育的保护作用。总而言之,我们通过改善突触和线粒体功能的损伤,降低tau磷酸化,Aβ1-42水平以及反应性氧种的产生,从而证明了N2A/APP细胞系中胃生育的神经保护作用。这些结果提供了对胃毒素对治疗阿尔茨海默氏病的潜在影响的新机械见解。
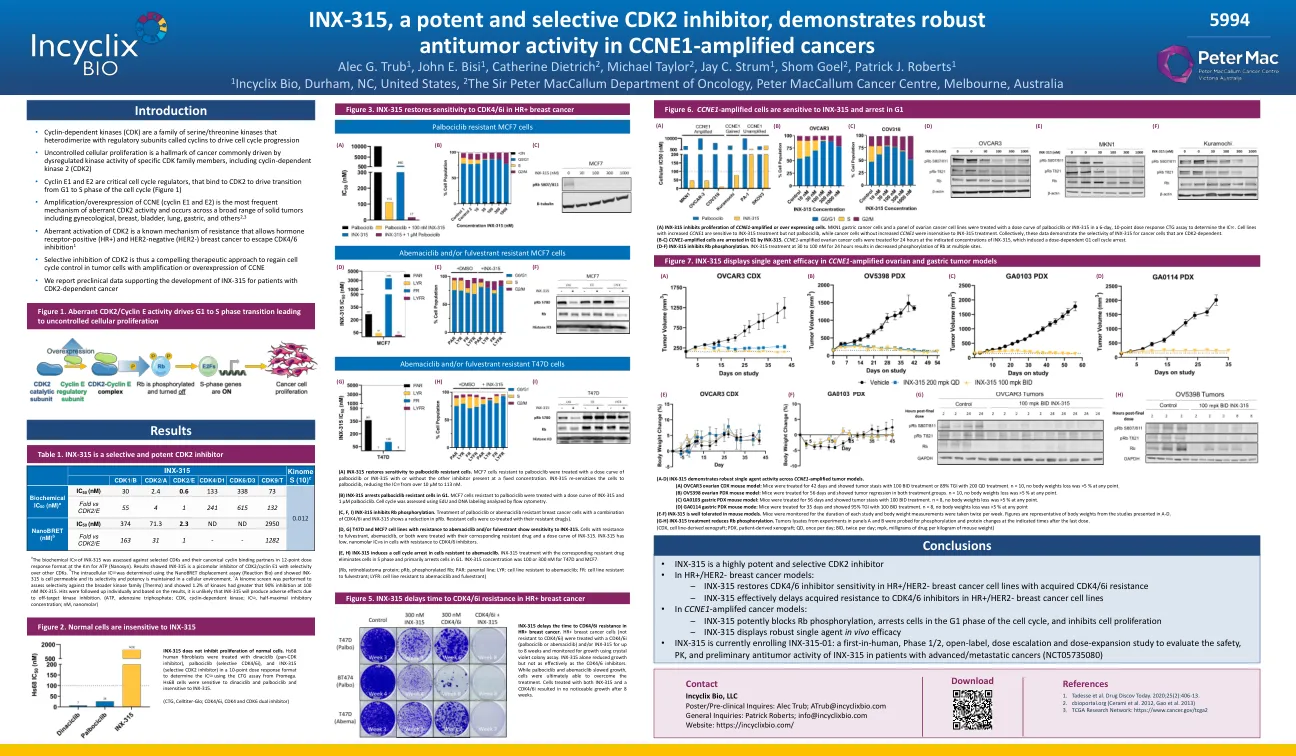
inx-315,一种有效的选择性CDK2抑制剂,证明了强大的
(a)INX -315抑制CCNE1扩增或超出表达细胞的增殖。MKN1胃癌细胞和一组卵巢癌细胞系在6天的10点剂量反应CTG分析中用palbociclib或INX-315的剂量曲线处理,以确定IC 50。CCNE1增加的细胞系对INX-315治疗敏感,而不是palbociclib,而没有增加CCNE1的癌细胞对INX-315治疗不敏感。 共同证明了INX-315对CDK2依赖性的癌细胞的选择性。 (B-C)CCNE1扩增细胞在G1中通过INX-315停止。 CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。 (D-F)INX-315抑制RB磷酸化。 INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。CCNE1增加的细胞系对INX-315治疗敏感,而不是palbociclib,而没有增加CCNE1的癌细胞对INX-315治疗不敏感。共同证明了INX-315对CDK2依赖性的癌细胞的选择性。(B-C)CCNE1扩增细胞在G1中通过INX-315停止。 CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。 (D-F)INX-315抑制RB磷酸化。 INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。(B-C)CCNE1扩增细胞在G1中通过INX-315停止。CCNE1-放大卵巢癌细胞在指定的INX-315浓度下处理24小时,这诱导剂量依赖性G1细胞周期停滞。(D-F)INX-315抑制RB磷酸化。INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。INX-315在30至100 nm的处理24小时下处理导致多个位点的RB磷酸化降低。

RPT6的磷酸化控制其结合DNA并调节在记忆形成期间雄性大鼠海马中基因表达的能力
研究文章| Cellular/Molecular Phosphorylation of RPT6 controls its ability to bind DNA and regulate gene expression in the hippocampus of male rats during memory formation https://doi.org/10.1523/JNEUROSCI.1453-23.2023 Received: 1 August 2023 Revised: 31 October 2023 Accepted: 29 November 2023 Copyright © 2023 the authors

p53 DNA结合合作的磷酸化控制平衡肿瘤发生和老化的Oleg timofeev 1,Lukas Koch 1,康斯坦丁NIEDERAU 1,ALI
摘要◥翻译后修饰对于调节转录因子p53至关重要,该转录因子p53以高度合作的方式结合DNA,以控制众多肿瘤抑制程序的表达。在这里,我们在DNA结合域中在高度保守的丝氨酸残基(人类S183/ S185,小鼠S180)的磷酸化中降低了DNA结合的合作性,从而显示了DNA结合的合作性。为探索这种抑制性磷酸化在体内的作用,生成了新的磷酸化 - 确定的p53-S180A敲入小鼠。染色质免疫沉淀测序和S180A敲入细胞的RNA测序研究表明DNA结合增强并增加了靶基因表达。在体内,这转化为骨髓的组织特异性脆弱性,导致造血干细胞的延伸,并损害DNA损伤后造血的适当再生。中位寿命显着从709天的野生型降低到仅568天

同工型和磷酸化特异性的多重多重定量药效学对靶向PI3K和MAPK信号传导的药物和临床活检
摘要◥ras/raf/mek/erk(MAPK)和pi3k/akt信号通路影响涉及癌症的几个细胞功能,使它们成为有吸引力的药物靶标。我们描述了一种新型的多重元素 - 用于定量PI3K/AKT和MAPK途径中蛋白质蛋白质的同工型特异性磷酸化,以评估小型动力学变化。在具有验证的抗体试剂验证的Luminex平台上开发了ERK1/2,MEK1/2,AKT1/2/3和RPS6的ERK1/2,MEK1/2,AKT1/2/3和RPS6的总蛋白质和特异性磷酸化水平的同工型特异性测定。多重分析表现出令人满意的分析性能。使用选定药物处理的异种移植模型进行拟合验证。在PC3和HCC70异种移植肿瘤中,PI3K B抑制剂AZD8186在单剂量后4至7小时抑制Akt1,Akt2和RPS6的磷酸化,但水平返回到