XiaoMi-AI文件搜索系统
World File Search System细胞系

单细胞分选,隔离和纳米素分配OMICS,稀有细胞和细胞系开发应用
如果下一个液滴仅包含一个适合用户定义的参数(大小,荧光标记)的单个单元格,则将其分配到目标实验室中。否则,将其分配到恢复瓶中,允许重新处理

临床前嗜铬细胞瘤和副神经节瘤模型_细胞系、动物模型和人类原代培养模型
尽管数十年的研究证明建立人类嗜铬细胞瘤和副神经节瘤 (PPGL) 细胞系尤为困难,但目前还有其他可靠的临床前 PPGL 模型可用。本综述总结了这些模型,以及我们最近使用患者来源的 PPGL 原代培养物建立的个性化药物筛选平台。当前可用的 PPGL 模型包括小鼠和大鼠 PPGL 细胞系(其中只有一个细胞系(PC12)可通过细胞库公开访问)和 PPGL 动物模型(其中患者来源的异种移植模型很有前景但建立起来很复杂)。我们已经开发出下一代人类 PPGL 原代培养物实施方案,能够根据肿瘤独特的遗传、生化、免疫组织化学和临床特征进行可靠和个性化的药物筛选,以及对肿瘤药物反应性的个体化分析。总体而言,可靠的 PPGL 模型(包括患者来源的原代培养模型)对于推进 PPGL 领域的临床前研究至关重要。© 2024 作者。由 Elsevier Ltd. 出版。这是一篇根据 CC BY 许可开放获取的文章(http://creativecommons.org/licenses/by/4.0/)。

研究二甲双胍和昔罗舍平对 PC3 和 THP-1 癌细胞系的合成致死率
摘要 口服抗糖尿病药二甲双胍已被证实在各种癌细胞系中具有抗肿瘤活性,与线粒体复合物 I 的抑制有关。然而,临床前研究一直难以证明这种抗肿瘤活性,其利用的二甲双胍浓度与标准抗糖尿病剂量可达到。研究表明,二甲双胍与抗高血压药物昔洛舍平联合使用,通过抑制单羧酸转运蛋白 (MCT)1/MCT4,可降低二甲双胍的治疗阈值并使癌细胞对杀伤敏感。这些药物之间的强相互作用可引起对转化细胞特有的合成致死性。这项初步研究旨在通过测量 PC3(粘附)和 THP-1(悬浮)癌细胞系中的细胞活力和细胞外乳酸来研究二甲双胍的抗肿瘤作用及其与昔洛舍平的协同关系。总体而言,PC3 细胞系对二甲双胍和昔罗舍平联合治疗或单独使用其中一种药物的治疗反应更好;然而,在两种细胞系中均未观察到合成致死。在两种细胞系中,二甲双胍和昔罗舍平之间的相互作用在细胞活力测定中并不具有统计学意义(p>0.05)。通过测量细胞外乳酸对 MCT1/MCT4 抑制的分析并不具有统计学意义(p>0.05),并且结果尚无定论。此外,在某些治疗组中,细胞系的性质(粘附或悬浮)具有统计学意义(p<0.05),这表明这可能在药物治疗的疗效中发挥作用。需要进一步研究以更好地了解二甲双胍和昔罗舍平合成致死以及昔罗舍平 MCT1/MCT4 抑制的潜在细胞机制。未来的研究应侧重于实现能够在体内表现出抗肿瘤作用的二甲双胍剂量。关键词:二甲双胍、昔罗舍平、THP-1、PC3、单羧酸转运蛋白、MCT1、MCT4、粘附细胞系、悬浮细胞系、前列腺癌、急性髓细胞白血病、抗肿瘤、合成致死。

反选择标记 PIGA 在工程设计缺失细胞系中的应用以及 CRISPR 缺失效率的表征
CRISPR / Cas9 系统是一种基因组工程技术,已应用于基因的插入/缺失突变以及靶基因的缺失和替换。在这里,我们描述了人类 X 染色体上 PIGA 基因座上的成对 gRNA 缺失,范围从 17 kb 到 2 Mb。我们发现缺失大小和缺失效率之间没有明显的线性相关性,拓扑关联域对缺失频率没有实质性影响。利用这种精确的缺失技术,我们设计了一系列设计缺失细胞系,包括一种缺失两个 X 染色体反选择(负选择)标记 PIGA 和 HPRT1 的细胞系,以及带有每个单独缺失的其他细胞系。PIGA 编码糖基磷脂酰肌醇 (GPI) 锚生物合成装置的组成部分。 PIGA 基因反选择标记具有独特的特点,包括现有的单细胞水平 PIGA 功能和功能丧失检测,以及存在一种有效的反选择剂 proaerolysin,我们经常用它来筛选表达 PIGA 的细胞。这些设计细胞系可以作为具有多种选择标记的通用平台,可能特别适用于大规模基因组工程项目,例如 Genome Project-Write (GP-write)。

建立永生海洋鱼细胞系作为体外工具,促进环境监测、生物技术和生物多样性的研究
永生化细胞系对研究人员来说非常宝贵,因为它们可以无限增殖,从而可以培养多代。与寿命有限的原代细胞不同,永生化细胞系(也称为连续细胞系)可以避免伦理问题、提取困难、传代能力有限以及由于细胞来源不同而导致的结果不一致等挑战。实验室条件下的大多数细胞都面临海弗利克极限,即端粒随着每次分裂而缩短,导致衰老。永生化细胞系克服了这些限制,可以进行稳定的长期研究,同时无需重复分离和培养细胞,从而节省了研究的时间和资源。连续细胞系可以在体外无限增殖,为广泛的研究目的提供可持续和可重复的系统。这些细胞系为在细胞和分子水平上研究生物体提供了一个独特的平台,提供了全生物模型并不总是能够提供的见解。它复制了宿主的细胞和遗传同质性,同时最大限度地减少了体内系统固有的变异性。因此,人们越来越关注开发新的细胞系以支持更广泛的生物学研究。

开发一种简单的培养方法,用于被微生物污染的组织,并应用于建立鱼类细胞系
细胞培养系统已用于研究遗传分析,激素调节,细胞因子分泌,病毒滴定和药物敏感性,以代替活动物,因为培养的细胞模仿了实验中的整个生物体。因此,将来将增加细胞培养系统的有用性。特别是,在细胞毒性化合物的assray中,不需要动物的系统非常出色。是从大鼠,小鼠和人类等乳腺组织中建立了大量细胞系,因为它们已在实验室中被用于实验室。此外,精确地研究了许多生化反应。最近,不仅从科学的角度,而且还从社会观察者那里讨论了环境激素(内部灌木丛)或二恶英对生物体的影响。要评估这些影响,还应检查其他动物,因为它们直接暴露于环境污染物。因此,鱼是研究这些综合对生物体影响的最好动物之一(Babich和Borenfreund,1987)。此外,许多来自g,鳍,性腺,睾丸,肾脏等的鱼类细胞系。(Wolf and Mann,1980;

如何将 IPSC 线路存入欧洲...
• 提高您的机构、研究和 iPSC 系的知名度。将您的细胞系存入 EBiSC,可使您的 iPSC 系和研究结果更加可见,因为这样可以让广大社区都能使用您的 iPSC。您的机构也可通过我们的标准化命名系统得到认可。 • 通过 EBiSC 传播细胞系。该库通过响应要求提供您的细胞系样本的研究人员并完成多项材料转让协议来节省您的时间。完成一次存入过程后,EBiSC 将完成其余工作。 • 备份存储。如果您的低温冷冻机出现故障或您本地持有的库存受到污染,您将有机会要求将您的细胞系样本退回实验室,只需支付运费。 • 保留自我分发的权利。通过您的细胞系存入,您不仅满足了资助机构和期刊共享已发表细胞系的要求,还保留了知识产权,并可以根据需要继续使用和分发它们。 • 与 EBiSC 合作伙伴合作。 EBiSC 由学术、中小企业和大型工业合作伙伴组成。通过成为 EBiSC 细胞系所有者,您将成为 EBiSC 内外未来潜在合作者的推荐人。• 额外的特性数据。EBiSC 将作为细胞系银行和质量控制程序的一部分执行额外的特性,这些数据将与您作为细胞系的存放者共享,以支持您正在进行的研究活动。• 商业授权。通过 EBiSC 传播您的细胞系可增加您的细胞系获得下游应用商业许可的机会,并使谈判和决策过程完全由您自行决定。• 免费访问 EBiSC iPSC 细胞系。对于每个存放的细胞系,您将免费收到 2 个小瓶,可以是 EBiSC 银行后的存放细胞系,也可以是另一个银行的 EBiSC 细胞系。*
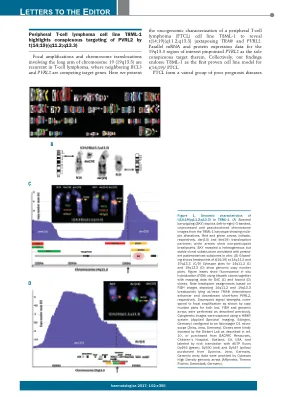
周围T细胞淋巴瘤细胞系T8ML-1突出显示了T(14; 19)(Q11.2; Q13.3)
图1。T(14; 19)(Q11.2; Q13.3)在T8ML-1中的基因组特征。 (a)光谱核分型(天空)描绘了来自T8ML-1核型的G带,未加工和伪色彩的染色体图像,显示了多个PLE改变。 红色和绿色箭头分别表示DER(14)和DER(19)易位伙伴;白色箭头显示非参与者断点。 天空揭示了与持续存在的患者衍生的亚克隆一致的异质但稳定的克隆下结构。 (b)G频段显示了14Q11.2和19Q13.3的T(14; 19)的断点。 (c/d)14q11.2(c)和19q13.3(d)的Cytoscan图显示了基因组拷贝数图。 图插图显示了使用Tilepath克隆以及映射BAC(C)和Fosmid(D)克隆的映射数据的荧光原位杂交(FISH)。 请注意基于鱼图像的断点分配,描绘了14q11.2和19q13.3分别位于Tra@ dowr@下游增强子和下游短形式PVRL2的断点。 差异信号强度符合焦点扩增,如两个基因座的拷贝数图所示。 如前所述,进行了鱼类和基因组阵列。 使用HISKY系统(Applied Spectral Imaging,Edingen,Germany)捕获了细胞遗传学图像,该系统配置为AxioImager D1 Micro-Scope(Zeiss,Jena,Germany)。 如参考文献中所述,Siebert Lab友好地捐赠了克隆。 10,或从美国加利福尼亚州奥克兰市的BACPAC资源,儿童医院购买,并由Nick Translation用Dutp Fluors Dy495(绿色),DY590(RED)和DY547(黄色)(黄色)(黄色)购买。T(14; 19)(Q11.2; Q13.3)在T8ML-1中的基因组特征。(a)光谱核分型(天空)描绘了来自T8ML-1核型的G带,未加工和伪色彩的染色体图像,显示了多个PLE改变。红色和绿色箭头分别表示DER(14)和DER(19)易位伙伴;白色箭头显示非参与者断点。天空揭示了与持续存在的患者衍生的亚克隆一致的异质但稳定的克隆下结构。(b)G频段显示了14Q11.2和19Q13.3的T(14; 19)的断点。(c/d)14q11.2(c)和19q13.3(d)的Cytoscan图显示了基因组拷贝数图。图插图显示了使用Tilepath克隆以及映射BAC(C)和Fosmid(D)克隆的映射数据的荧光原位杂交(FISH)。请注意基于鱼图像的断点分配,描绘了14q11.2和19q13.3分别位于Tra@ dowr@下游增强子和下游短形式PVRL2的断点。差异信号强度符合焦点扩增,如两个基因座的拷贝数图所示。鱼类和基因组阵列。使用HISKY系统(Applied Spectral Imaging,Edingen,Germany)捕获了细胞遗传学图像,该系统配置为AxioImager D1 Micro-Scope(Zeiss,Jena,Germany)。如参考文献中所述,Siebert Lab友好地捐赠了克隆。10,或从美国加利福尼亚州奥克兰市的BACPAC资源,儿童医院购买,并由Nick Translation用Dutp Fluors Dy495(绿色),DY590(RED)和DY547(黄色)(黄色)(黄色)购买。基因组阵列数据由Cytoscan高密度基因组阵列(Affymetrix,Thermo Fischer,Darmstadt,Germany)提供。

烟草悬浮细胞系中随机和靶向转基因整合事件之间的基因表达变异
摘要 锌指核酸酶等基因组编辑工具为植物的遗传操作提供了新策略。与农杆菌介导或直接基因转移不同,农杆菌介导或直接基因转移将基因随机引入基因组,从而可能导致基因表达的高度变异,而靶向基因添加可将 DNA 序列可预测地整合到植物基因组的特定位置。我们研究了各种独立的细胞系是否都含有通过锌指核酸酶介导的同源重组 (HR) 放置在相同基因组位点的转基因,与通过农杆菌介导的随机位点转化产生的整合事件相比,它们会产生更可重复和更均匀的表达水平。通过流式细胞术测量转基因产生的蛋白质量,分析了 Nicotiana tabacum L cv. Bright Yellow 2 (BY-2) 悬浮细胞中靶向 HR 事件和随机整合事件的基因表达差异,从而首次提供了关于快速增殖的植物悬浮细胞系中标记基因表达的位置效应的报告。靶向 HR 和单拷贝随机事件的标记蛋白水平覆盖了相似的范围;但是,给定细胞系中靶向事件的蛋白质表达均匀性明显高于随机插入转基因的细胞系;HR 细胞系原生质体的整体生存力也是如此。总之,使用靶向插入到已充分表征的细胞系的合格基因座中比随机插入到基因组中可获得更可靠的结果。

抗死亡通路诱导作为 CRISPR/Cas-9 敲除结肠直肠癌细胞系的潜在靶向治疗
受调节的细胞死亡是一种基本的生物学过程,在维持组织稳态和消除受损或不必要的细胞方面起着至关重要的作用。铁死亡是一种铁依赖性过程,特征是氧化和受损脂质的积累,从而导致程序性细胞死亡。在调节这一过程的铁死亡途径基因中,可以考虑GPX4、TFRC、ACSL4、FSP1、SLC7A11 和 PROM2。有许多众所周知的铁死亡途径调节剂,本综述将对此进行讨论。不同组织来源的细胞对这些调节剂表现出敏感或抗性表型。在某些情况下,细胞治疗过程中会发生意外变化,表明可能存在调节死亡途径。我们假设细胞(尤其是结直肠癌细胞系)从铁敏感性转变为铁抗性可能是诱导化学抗性的结果。利用 CRISPR/Cas-9 基因组编辑等新技术,可以实现诱导表型“转换”。