XiaoMi-AI文件搜索系统
World File Search System读取


人工智能甚至可以准确读取不常见的手写字符! !
*要使用此服务,您需要一个将文档数字化的设备,例如扫描仪和互联网连接。 ※本服务利用了AI inside Inc. 的字符识别AI。 ※我们与东日本电信电话株式会社签署了合作协议,并推出了手写字符读取服务。
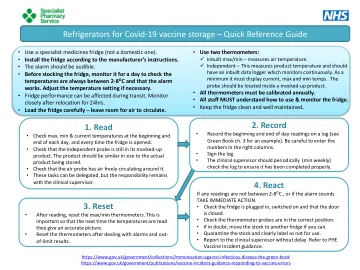
1. 读取 2. 记录 3. 重置 4. 对 Covid 冰箱做出反应...
4. 反应 如果任何读数不在 2-8 OC 之间,或者警报响起,请立即采取行动。 • 检查冰箱是否插上电源、打开电源并且门是否关闭。 • 检查温度计探头是否处于正确位置。 • 如有疑问,请尽可能将库存移至另一台冰箱。 • 隔离库存并明确标记为不可使用。 • 立即向临床主管报告。请参阅 PHE 疫苗事件指南。

128Gb 1 位/单元 96 字线层 3D 闪存可改善随机读取延迟,tProg = 75 μs 和 tR = 4 μs
摘要 — 采用 96 字线层技术开发了一款 128 Gb 1 位/单元 3-D 闪存芯片。一种具有较少字线和位线时间常数的新型芯片布局结构实现了快速读取访问时间。新引入的程序序列即使在写入/擦除循环后也能实现更高的可靠性和更少的读取重试。还采用了外部 VPP 电源 (12 V)、电流模式参考分布和自动温度代码刷新来提高芯片的性能。新的占空比校正器成功获得了更宽的 DQS 单位间隔。因此,所提出的芯片具有 4 µ s 的读取访问延迟和 75 µ s 的编程时间,比采用相同技术的传统 3-D 闪存快 12-13 倍和 4-5 倍 [Maejima et al. , (2018)]。随机读取延迟(tRRL)估计小于 50 µ s,这使得能够减少固态硬盘(SSD)系统的总读取访问时间。

基于多层 RRAM 的深度学习推理引擎的读取扰动和双极读取方案研究
摘要——基于多层电阻式随机存取存储器 (RRAM) 的突触阵列可以实现矢量矩阵乘法的并行计算,从而加速机器学习推理;然而,由于模拟电流沿列相加,因此单元的任何电导漂移都可能导致推理精度下降。在本文中,在基于 2 位 HfO 2 RRAM 阵列的测试车辆上统计测量了读取干扰引起的电导漂移特性。通过垂直和横向细丝生长机制对四种状态的漂移行为进行了经验建模。此外,提出并测试了一种双极读取方案,以增强对读取干扰的恢复能力。建模的读取干扰和提出的补偿方案被纳入类似 VGG 的卷积神经网络中,用于 CIFAR-10 数据集推理。

使用超低噪声 SQUID 放大器实现量子点的灵敏射频读取
半导体中的电子自旋是最先进的量子比特实现方式之一,也是利用工业工艺制造的可扩展量子计算机的潜在基础 [1–3]。一台有用的计算机必须纠正计算过程中不可避免地出现的错误,这需要很高的单次量子比特读出保真度 [4]。用于错误检测的全表面码要求在计算机的每个时钟周期内读出大约一半的物理量子比特 [5]。直到最近,自旋量子比特装置中的单次读出只能通过自旋到电荷的转换来实现,由附近的单电子晶体管 (SET) 或量子点接触 (QPC) 电荷传感器检测 [6–9]。然而,如果使用色散读出,硬件会更简单、更小,这利用了双量子点中单重态和三重态自旋态之间的电极化率差异 [10–13]。可以通过与量子点电极之一粘合的射频 (RF) 谐振器监测由此产生的两个量子比特状态之间的电容差异。量子点中的电荷跃迁也会发生类似的色散偏移,这样反射信号有助于调整到所需的电子占据 [14–16]。色散读出的优势在于它不需要单独的电荷传感器,但即使在自旋衰减时间较长的系统中,电容灵敏度通常也不足以进行单次量子比特读出 [17–23]。最近,已经在基于双量子点的系统中展示了色散单次读出 [24–28],但为了提高读出保真度,仍然需要更高的灵敏度。

嵌入式参考电极用于潜在读取(ere 20 ...
基于经过验证的电池技术,ERE 20是一个真正的半细胞,它使用钢壳中的锰二氧化碳电极和碱性无氯 - 无凝胶。钢壳由耐腐蚀材料制成。凝胶的pH值对应于正常混凝土中孔隙水的pH,因此消除了由于离子通过多孔塞扩散引起的误差。

通过体外转录实现 DNA 条形码的原位读取和单碱基编辑
唯一识别单个细胞的分子条形码技术受到条形码测量限制的阻碍。通过测序读取不会保留组织中细胞的空间组织,而成像方法保留了空间结构,但对条形码序列不太敏感。在这里,我们介绍了一种基于图像读取短(20bp)DNA条形码的系统。在这个称为Zombie的系统中,噬菌体RNA聚合酶在固定细胞中转录工程条形码。随后通过荧光原位杂交检测所得RNA。使用竞争匹配和错配探针,Zombie可以准确区分条形码中的单核苷酸差异。该方法允许原位读取密集的组合条形码库和由CRISPR碱基编辑器产生的单碱基突变,而无需在活细胞中表达条形码。Zombie可在多种环境中发挥作用,包括细胞培养、鸡胚和成年小鼠脑组织。通过成像灵敏地读取紧凑和多样化的DNA条形码的能力将促进广泛的条形码和基因组记录策略。

远程感知可以将电池充电时间减少50%,28SI配备了远程感知,该感官使用了第二条电线,该电线读取实际电压a
远程感知可以将电池充电时间减少50%。28SI配备了远程感知,该感觉利用了第二条电线,该电线读取电池处的实际电压。这标志着交流发电机以增加输出以补偿电压降,从而确保电池处的14伏。适当的电压力更多地进入电池,使其达到车辆停靠之间的全部充电状态。

iclass SE多类读取器RP10(900p)-Securitas
Typical Read Range iCLASS SE®: 6.4 cm, SE for DESFire® EV1: 2.5 cm, SE for MIFARE® Classic: 5.8 cm (13.56 MHz Single Technology ID-1 Credentials (Cards) – SIO Model Data) iCLASS SE: 2.5 cm, SE for MIFARE Classic: 1.3 cm (13.56 MHz Single Technology Tags/Fobs – SIO Data Model) HID Prox / AWID: 5.1 cm, Indala Prox: 2.5 cm, EM4102: 8.9 cm (125 kHz Single Technology ID-1 Credentials (Cards) – Respective Prox Data Model) HID Prox / AWID: 2.5 cm, Indala Prox: 1.3 cm, EM4102: 3.3 cm (125 KHz Single Technology Tags/Fobs – Respective Prox Data Model)