XiaoMi-AI文件搜索系统
World File Search System集合

从前额叶集合解码意识的内容
预印本(未经同行评审认证)是作者/资助者。保留所有权利。未经许可不得重复使用。此版本的版权所有者于 2020 年 1 月 28 日发布。;https://doi.org/10.1101/2020.01.28.921841 doi:bioRxiv 预印本

这是生活中最重要的事物的集合。
STIFA 继承了其前身——科学技术发展局(STDF),其具体目标是改善埃及的研发环境、资助科技活动和发展埃及的创新能力。STDF 在国家科技发展战略的背景下实施其目标,该战略由高级科学技术委员会制定。对埃及国民经济影响最大的埃及各部委在委员会中设有代表,负责指导研究活动,使其转向对国家发展计划有直接影响的科技活动。

集合数据生成的生成无序流量
基于流量的生成模型已经证明了广泛的数据模式(例如图像和文本)的有希望的性能。但是,很少有工作探索其扩展到无序数据(例如,空间点集),这并不是很微不足道,因为以前的模型主要是为自然订购的向量数据设计的。在本文中,我们提出了无序的流,这是一种基于流程的基于设定数据生成的生成模型。具体来说,我们将未订购的数据转换为适当的函数代表,并通过功能值流量匹配来了解此类表示的概率度量。对于从函数表示到未排序数据的逆映射,我们提出了一种类似于粒子过滤的方法,Langevin Dynamics首先要热身初始粒子和基于梯度的搜索,以更新它们直至结合。我们已经在多个现实世界数据集上进行了广泛的实验,这表明我们的无序流模型在生成集合结构化数据方面非常有效,并且显着胜过先前的基线。

基因库一个DNA库是DNA片段的集合...
纯化mRNA后,将寡-DT(脱氧 - 胸腺苷核苷酸的短序列)标记为互补底漆,该引物与poly-A尾巴结合,从而可以通过反转录酶来扩展自由3'-OH端,以创建互补的DNA链。现在,使用RNase酶去除去mRNA,将单个链cDNA(SSCDNA)取出。借助于DNA聚合酶,将此SSCDNA转化为双链DNA。但是,要使DNA聚合酶合成互补链,需要3'- oh-end。这是由SSCDNA本身通过在3'端产生发夹循环来通过围绕自身来提供的。聚合酶延伸了3'-OH端,然后通过S 1核酸酶的剪刀作用打开3'端的环。然后,使用限制性核酸内切酶和DNA连接酶来克隆序列到细菌质粒中。

fMRI大脑网络的微型典型和规范集合
重复使用本文是根据创意共享属性 - 非商业 - 诺迪维斯(CC BY-NC-ND)许可证的条款分发的。此许可只允许您下载此工作并与他人共享,只要您归功于作者,但是您不能以任何方式更改文章或商业使用。此处的更多信息和许可证的完整条款:https://creativecommons.org/licenses/

使用集合机学习技术的糖尿病预测
D. Madhu Sudhana Rao,D。Sai。Sridhathri信息技术部,Vignan科学技术与研究基金会,印度Guntur,Madhudontha@gmail.com,sridhathri.1021@gmail.com摘要。 糖尿病最称为糖尿病是一种急性内分泌慢性疾病,这是许多人通过遗传性或从人类生活方式的趋势中的主要问题。 由于内分泌问题,它会提高体内的血糖。 血糖的这种增加不仅会影响其水平,甚至会导致许多与肾脏,肝功能,血压和眼睛损害相关的健康问题。 这在较小的年龄组中最常见,对于45岁以上的年龄组。 我国将近68%的人患有这些糖尿病患者。 当预测靠近水平时,可以避免或消除它。 在情况下,它被认为存在严重的问题,并且需要以任何代价控制它。 结合了计算机科学的技术,我们使用机器学习技术在早期阶段预测糖尿病,以更高的准确性。 在这里,我们使用不同的分类器,即k-nearest,幼稚的贝叶斯(NB),XG提升,决策树(DT)和随机森林(RF),并从提供的数据集中检测其准确性。 与其他不同技术相比,我们发现随机森林更适合于更高的精度计算。Sridhathri信息技术部,Vignan科学技术与研究基金会,印度Guntur,Madhudontha@gmail.com,sridhathri.1021@gmail.com摘要。糖尿病最称为糖尿病是一种急性内分泌慢性疾病,这是许多人通过遗传性或从人类生活方式的趋势中的主要问题。由于内分泌问题,它会提高体内的血糖。血糖的这种增加不仅会影响其水平,甚至会导致许多与肾脏,肝功能,血压和眼睛损害相关的健康问题。这在较小的年龄组中最常见,对于45岁以上的年龄组。我国将近68%的人患有这些糖尿病患者。当预测靠近水平时,可以避免或消除它。在情况下,它被认为存在严重的问题,并且需要以任何代价控制它。结合了计算机科学的技术,我们使用机器学习技术在早期阶段预测糖尿病,以更高的准确性。在这里,我们使用不同的分类器,即k-nearest,幼稚的贝叶斯(NB),XG提升,决策树(DT)和随机森林(RF),并从提供的数据集中检测其准确性。与其他不同技术相比,我们发现随机森林更适合于更高的精度计算。
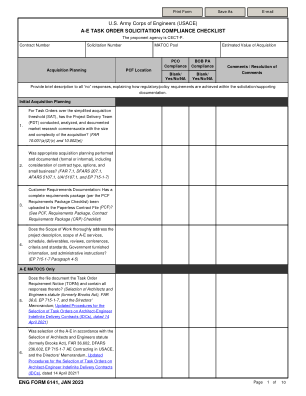
AE 任务订单征集合规检查表
ACO - 行政合同官 AE - 建筑工程师 AFARS - 陆军联邦采购条例补充 AT/OPSEC - 反恐行动安全 BCOES - 可投标性、可施工性、可操作性、环境和可持续性 BOB - 业务监督部门 CAR - 合同行动报告 CCC - 中心合同主管 CoCO - 合同办公室主任 CLINS - 合同项目编号 COR - 合同官员代表 CPARS - 承包商绩效评估报告系统 CRB - 合同审查委员会 D&Fs - 决心与发现 DFARS - 国防联邦采购条例补充 DOC - 合同主管 DoD SSP - 国防部源选择程序 EVMS - 挣值管理系统 FAR - 联邦采购条例 HCA - 合同活动主管

使用集合机学习技术的糖尿病预测
D. Madhu Sudhana Rao,D。Sai。Sridhathri信息技术部,Vignan科学技术与研究基金会,印度Guntur,Madhudontha@gmail.com,sridhathri.1021@gmail.com摘要。 糖尿病最称为糖尿病是一种急性内分泌慢性疾病,这是许多人通过遗传性或从人类生活方式的趋势中的主要问题。 由于内分泌问题,它会提高体内的血糖。 血糖的这种增加不仅会影响其水平,甚至会导致许多与肾脏,肝功能,血压和眼睛损害相关的健康问题。 这在较小的年龄组中最常见,对于45岁以上的年龄组。 我国将近68%的人患有这些糖尿病患者。 当预测靠近水平时,可以避免或消除它。 在情况下,它被认为存在严重的问题,并且需要以任何代价控制它。 结合了计算机科学的技术,我们使用机器学习技术在早期阶段预测糖尿病,以更高的准确性。 在这里,我们使用不同的分类器,即k-nearest,幼稚的贝叶斯(NB),XG提升,决策树(DT)和随机森林(RF),并从提供的数据集中检测其准确性。 与其他不同技术相比,我们发现随机森林更适合于更高的精度计算。Sridhathri信息技术部,Vignan科学技术与研究基金会,印度Guntur,Madhudontha@gmail.com,sridhathri.1021@gmail.com摘要。糖尿病最称为糖尿病是一种急性内分泌慢性疾病,这是许多人通过遗传性或从人类生活方式的趋势中的主要问题。由于内分泌问题,它会提高体内的血糖。血糖的这种增加不仅会影响其水平,甚至会导致许多与肾脏,肝功能,血压和眼睛损害相关的健康问题。这在较小的年龄组中最常见,对于45岁以上的年龄组。我国将近68%的人患有这些糖尿病患者。当预测靠近水平时,可以避免或消除它。在情况下,它被认为存在严重的问题,并且需要以任何代价控制它。结合了计算机科学的技术,我们使用机器学习技术在早期阶段预测糖尿病,以更高的准确性。在这里,我们使用不同的分类器,即k-nearest,幼稚的贝叶斯(NB),XG提升,决策树(DT)和随机森林(RF),并从提供的数据集中检测其准确性。与其他不同技术相比,我们发现随机森林更适合于更高的精度计算。

在量子旋转集合中长寿的挤压基态
我们在原子旋转1玻色的凝结物中产生自旋挤压基态,该凝结物在量子临界点附近调节,该量子使用一种新型的非绝热技术将相互作用集合的不同自旋阶段分开。与典型的非平衡方法相反,用于通过量子相变的淬灭原子挤压状态,挤压的基态是及时的,具有恒定的正交挤压角。挤压的基态有6-8 dB的挤压和恒定的挤压角。测量挤压基态的长期演变,并显示出2 s的挤压程度的逐渐下降,这是由于原子密度损失而通过缓慢调整汉密尔顿的良好模型。有趣的是,尽管损失了75%的原子,但对逐渐减小的建模不需要额外的自旋脱碳模型。

使用CNN集合模型基于脑电图的重叠划分...
※这是根据创意共享归因非商业许可(http://creativecommons.org/licenses/by-nc/3.0/)发行的开放访问文章,允许在任何媒介中在任何媒介中进行无限制的非商业用途,分发和复制,前提是原始工作是正确的。