XiaoMi-AI文件搜索系统
World File Search SystemGBA1

GBA1突变改变了...
(未通过同行评审认证)是作者/资助者。保留所有权利。未经许可就不允许重复使用。此预印本版的版权持有人于2024年8月7日发布。 https://doi.org/10.1101/2024.08.05.606574 doi:biorxiv Preprint

GBA1会议,6月27日至29日,加拿大蒙特利尔GBA1会议,6月27日至29日,加拿大蒙特利尔
15:30 – 15:42 Deciphering the Immunogenomic Landscape of GBA1-Associated Parkinson's Disease – Towfique Raj 15:42 – 15:54 The role of the ‘African GBA1 allele' of rs3115534 in Parkinson's Disease – Peter Bauer 15:54 – 16:06 Acid ceramidase plays a key role in the pathogenic cascade leading to neurodegeneration in Gaucher and GBA1-associated Parkinson's disease – Ricardo A. Feldman 16:06 – 16:18 A Genome Wide CRISPR Interference Screen Reveals Commander Genes as Modifiers of Glucocerebrosidase Activity and Lysosomal Function – Nathaniel Safren 16:18 – 16:30 A Genome-Wide Landscape of Genetic Modifiers of GCase Deficiency Revealed by An Arrayed CRISPR激活屏幕 - 江-An yin 16:30 - 16:42加剧的溶酶体和线粒体功能障碍与GBA1的常见帕金森氏病风险等位基因相关 - Oliver B. Davis 16:42 - 16:54在GBA1-PARKOLISS ON GABA1-PARSISSON SISTIS-PRIPALISM中的性中断 - <
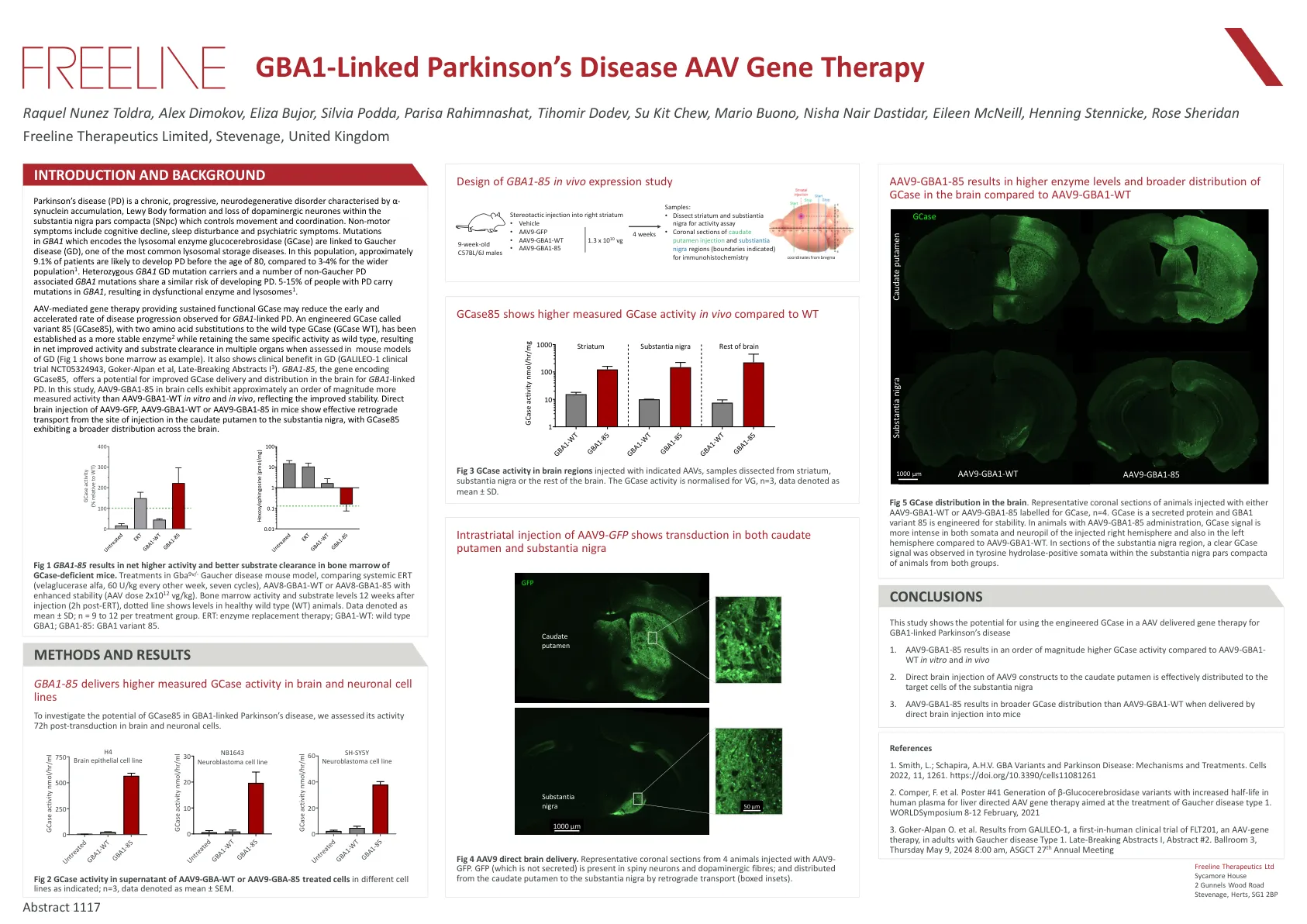
与GBA1连接的帕金森氏病AAV基因治疗
帕金森氏病(PD)是一种慢性,进行性,神经退行性疾病,其特征是α-突触核蛋白积累,Lewy身体形成和质体nigra Pars Compacta(SNPC)中多巴胺能神经元的丧失,控制运动和配位。非运动症状包括认知能力下降,睡眠障碍和精神病症状。GBA1中编码溶酶体酶葡萄糖脑苷酶(GCASE)的突变与Gaucher病(GD)有关,Gaucher病(GD)是最常见的溶酶体储存疾病之一。在这一人群中,大约9.1%的患者可能在80岁之前发展PD,而更广泛的人群为3-4%。杂合GBA1 GD突变携带者和许多非Gaucher PD相关的GBA1突变具有相似的发展PD风险。5-15%的PD患者在GBA1中携带突变,导致功能失调的酶和溶酶体1。

α-突触核蛋白内含子 1 的 DNA 甲基化显著...
摘要:帕金森病 (PD) 是一种常见的运动障碍,估计到 80 岁为止,有 4% 的人会患有此病。葡萄糖脑苷脂酶 1 (GBA1) 基因突变是 PD 最常见的遗传风险因素,至少 7-10% 的非德系 PD 个体携带 GBA1 突变 (PD-GBA1)。尽管与特发性 PD 相似,但 PD-GBA1 的临床表现包括发病年龄略低、神经精神症状发生率更高,并且认知障碍往往更早、更普遍且更严重。PD-GBA1 的病理生理机制尚不完全清楚,但与特发性 PD 一样,α-突触核蛋白积累被认为起着关键作用。有人假设这种 α-突触核蛋白的过度表达是由表观遗传修饰引起的。在本文中,我们分析了特发性 PD、PD- GBA1 和老年非 PD 对照者三个不同脑区(额叶皮质、壳核和黑质)中内含子 1 和 α -突触核蛋白 ( SNCA ) 基因启动子内的 17 个 CpG 位点的 DNA 甲基化水平。在这三个脑区中,我们发现特发性 PD 和 PD- GBA1 的内含子 1 的 8 个 CpG 区域内的 DNA 甲基化呈下降趋势。DNA 甲基化降低的趋势在 PD- GBA1 中更为明显,额叶皮质的下降更为显著。这表明 PD- GBA1 和特发性 PD 具有不同的表观遗传特征,并强调了区分特发性 PD 和 PD- GBA1 病例的重要性。这项工作还提供了初步证据,表明 PD 中可能存在不同的遗传亚型,每种亚型都有其自身的病理机制。这可能对 PD 的诊断和治疗方式具有重要意义。

开发AAV-GBA1基因替代疗法,用于通过血液屏障渗透AAV CAPSID
a-b)VG生物分布是通过DDPCR,FAM-RBGPA和VIC-MSTFRC探针来测量二倍体动物细胞的。在皮质(CTX)和丘脑(Th)中证明了跨前脑和中脑区域的成功基因转移。最小基因转移(<1VG/细胞)。d -f)GBA1 mRNA表达。用于该测定法的7个PLEX探针集是定制的,旨在区分转基因特异性GBA mRNA和小鼠内源性GBA mRNA。mRNA表达值据报道为平均荧光强度(MFI)。人类GBA1 mRNA如图d -f所示。在皮质和丘脑中证明了跨大脑区域的成功转录。在肝脏中观察到降低的表达。g - i)使用Sensolote®蓝葡萄糖脑培合酶活性测定法测量皮质,丘脑和肝脏的Gcase活性。高剂量的Php.eb.gba1具有增强10、2、5和3的高剂量,在中枢神经系统组织中的GCASE活性显着增加。平均值+/- SEM。

早期诊断Gaucher病的障碍
摘要:Gaucher疾病(GD)是一种罕见的溶酶体储存障碍,是由于GBA1基因中双质体变体引起的溶酶体酶葡萄糖脑苷酶的缺乏。患者可能会出现各种各样的疾病表现,包括肝脑膜全脂,血小板减少症,骨骼表现,以及GD类型2和3型,神经退行性变性,认知延迟和/或眼球运动异常。尽管没有治疗神经性GD的治疗方法,但可以通过酶替代疗法或底物还原疗法有效地进行非神经性表现。但是,许多GD患者经历了长时间的诊断性奥德赛,这可能会对他们的护理和临床结果产生负面影响。此诊断延迟的原因是多方面的。由于GD中的基因型/表型相关性并不总是很清楚,因此很难预测临床表现的存在,严重程度和发作。这种异质性,结合了GBA1基因座的分子复杂性,低疾病患病率以及对提供者的GD知识有限,这是GD早期诊断的障碍。在这篇评论中,我们讨论了改善GD患者诊断旅程的障碍和挑战,考虑因素和未来步骤。关键字:Gaucher病,新生儿筛查,诊断

溶酶体存储,高彻和帕金森氏病的自噬和先天免疫受损:药物发现的见解
自噬 - 溶酶体途径的损害越来越涉及帕金森氏病(PD)。GBA1突变引起溶酶体储存障碍Gaucher病(GD),是PD的最常见遗传危险因素。GBA1突变已显示会引起自噬 - 溶酶体损伤。 不良细胞成分的自噬降解有缺陷与多种病理有关,包括正常蛋白质稳态的丧失,特别是α-突触核蛋白和先天免疫功能障碍。 在PD和GD中观察到后者。 在这里,我们将讨论自噬和免疫失调之间的机理联系,以及这些病理学在肠道和大脑之间在这些疾病中的沟通中的可能作用。 在神经性GD(NGD)的蝇模型中的最新工作显示肠自噬缺陷导致胃肠道功能障碍和免疫激活。 雷帕霉素治疗部分逆转了自噬阻滞并降低了免疫活性,与生存率增加并改善了运动能力。 肠道微生物组的改变是神经炎症的关键驱动力,研究表明,在NGD蝇中消除了微生物组,而PD的小鼠模型可以改善脑部炎症。 在这些观察结果之后,将溶酶体 - 自噬途径,先天免疫信号传导和微生物组营养不良症讨论为PD和GD中的潜在治疗靶标。 本文是讨论会议问题的一部分,“理解神经变性中的内聚糖网络”。GBA1突变已显示会引起自噬 - 溶酶体损伤。不良细胞成分的自噬降解有缺陷与多种病理有关,包括正常蛋白质稳态的丧失,特别是α-突触核蛋白和先天免疫功能障碍。在PD和GD中观察到后者。在这里,我们将讨论自噬和免疫失调之间的机理联系,以及这些病理学在肠道和大脑之间在这些疾病中的沟通中的可能作用。在神经性GD(NGD)的蝇模型中的最新工作显示肠自噬缺陷导致胃肠道功能障碍和免疫激活。雷帕霉素治疗部分逆转了自噬阻滞并降低了免疫活性,与生存率增加并改善了运动能力。肠道微生物组的改变是神经炎症的关键驱动力,研究表明,在NGD蝇中消除了微生物组,而PD的小鼠模型可以改善脑部炎症。在这些观察结果之后,将溶酶体 - 自噬途径,先天免疫信号传导和微生物组营养不良症讨论为PD和GD中的潜在治疗靶标。本文是讨论会议问题的一部分,“理解神经变性中的内聚糖网络”。

神经退行性疾病进入功能基因组学时代
低成本 DNA 测序的普及使另一种将人类遗传学与神经系统疾病的特定基因驱动因素联系起来的方法——全基因组关联研究 (GWAS) 复活。通过比较患病个体与未患病(“对照”)人群的基因组成,可以确定增加患病可能性的风险因素。GWAS 导致发现特定基因的变异,包括 TREM2(髓系细胞 2 上表达的触发受体)和 GBA1(葡萄糖脑苷脂酶 1),分别是非孟德尔 AD 和 PD 的风险因素。在某些情况下,GWAS 结果突出了以前被低估的导致发病的机制。例如,与 AD 风险相关的遗传变异在髓系细胞(可能是小胶质细胞)中起作用的基因和增强子(基因组中控制基因表达的区域)中富集。这表明先天免疫细胞在 AD 中发挥着重要作用。因此,特定生物途径中风险变异的丰富可以加深我们对神经退行性疾病的机制理解,甚至可能指出新的治疗目标。
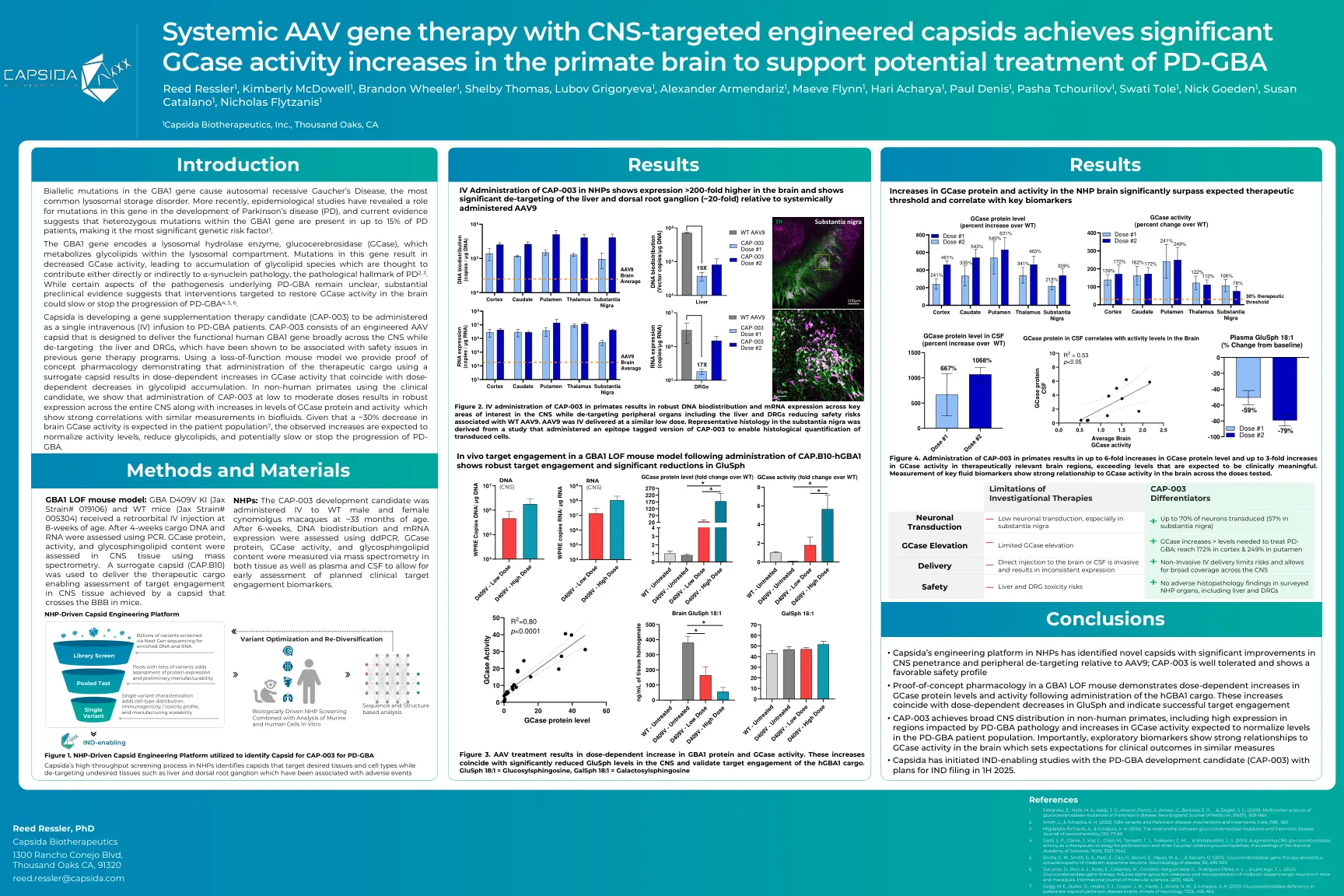
具有CNS靶向工程的Capsids的全身性AAV基因治疗可实现灵长类动物大脑的显着gcase活性,以支持潜在的
Capsida正在开发一种基因补充疗法候选者(CAP-003),该疗法被作为单一静脉注射(IV)输注对PD-GBA患者。CAP-003由一个工程的AAV CAPSID组成,旨在在CNS上广泛提供功能性的人GBA1基因,同时对肝脏和DRG进行靶向,这些基因已被证明与以前的基因治疗计划中的安全问题有关。使用功能丧失的小鼠模型,我们提供了概念药理学证明,表明使用替代capsid的治疗货物的给药会导致GCASE活性的剂量依赖性增加,而GCASE活性与糖脂积累的剂量依赖性降低相吻合。在非人类灵长类动物中使用临床候选者,我们表明,在低至中剂量下的CAP-003给药会导致整个CNS的稳健表达,同时GCASE蛋白水平和活性的增加,这表明与生物含量相似的测量相关性很强。鉴于患者人群的脑gcase活性降低了约30%,预计观察到的增加将使活性水平归一化,降低糖脂,并可能降低或停止PD-GBA的进展。

碳水化合物代谢遗传疾病中的代谢性心肌病和心脏缺陷:系统评价。 Conte,F。; Sam,J.E。; Lefeber,
摘要:心力衰竭(HF)是一种进行性慢性病,仍然是全球死亡的主要原因,影响了6400万以上的患者。HF可能是由具有单基因病因的心肌病和先天性心脏缺陷引起的。与心脏缺陷发展相关的基因和单基因疾病的数量正在不断增长,并包括遗传的代谢杂志(IMD)。已经报道了几种影响各种代谢途径的IMD,出于心肌病和心脏缺陷。考虑到糖代谢在心脏组织中的关键作用,包括能量产生,核酸合成和糖基化,与心脏表现相关的越来越多的与碳水化合物代谢相关的IMD越来越多。在这项系统的综述中,我们提供了与碳水化合物代谢相关的IMD的全面概述,这些IMD呈现出心肌病,心律失常疾病和/或结构性心脏缺陷。我们识别出患有心脏并发症的58 IMD:3糖/糖连接转运蛋白的缺陷(GLUT3,GLUT10,THTR1); 2个磷酸盐途径的疾病(G6PDH,TALDO); 9糖原代谢疾病(GAA,GBE1,GDE,GYG1,GYS1,LAMP2,RBCK1,PRKAG2,G6PT1); 29 congenital disorders of glycosylation (ALG3, ALG6, ALG9, ALG12, ATP6V1A, ATP6V1E1, B3GALTL, B3GAT3, COG1, COG7, DOLK, DPM3, FKRP, FKTN, GMPPB, MPDU1, NPL, PGM1, PIGA, PIGL, PIGN, PIGO,PIGT,PIGV,PMM2,POMT1,POMT2,SRD5A3,XYLT2); 15碳水化合物连接的溶酶体储存疾病(CTSA,GBA1,GLA,GLB1,HEXB,IDUA,IDS,IDS,SGSH,NAGLU,HGSNAT,GNS,GNS,GALNS,GALNS,GALNS,ARSB,ARSB,GUSB,GUSB,ARSK)。通过这项系统评价,我们旨在提高人们对碳水化合物连接IMD的心脏介绍的认识,并引起人们对碳水化合物连接的致病机制的注意,这些致病机制可能是心脏并发症的基础。