XiaoMi-AI文件搜索系统
World File Search SystemHSPC

急性髓样白血病进展过程中骨髓微环境的重塑
急性髓样白血病(AML)由恶性造血茎和祖细胞(HSPC)克隆造成正常造血的劫持。根据美国癌症学会的说法,AML是成年人口中第二大常见的白血病类型,占所有白血病病例的31%。5年生存率小于28%的AML患者的预后率显着较低(1)。尽管在治疗AML方面取得了很多进展,但这些疗法无法完全治愈该疾病。疾病复发很大程度上是由于白血病干细胞(LSC)从化学治疗药物和抗癌药中逃脱的。骨髓(BM)小裂被认为是通过BM微环境细胞与白血病细胞之间的双向相互作用而重塑的,以偏向AML的进展(2)。在白血病发生期间,BM生态位的恶化也会提高AML-HSPC和白血病爆炸增殖之间的竞争力(3)。理解正常造血过程中各种造血干细胞(HSC) - 氮相互作用变得很重要。进一步了解白血病期间BM壁ches重塑的动力学构成了现代癌症研究的组成部分。针对这些白血病壁ni,正在成为开发新型治疗AML治疗策略的新途径。
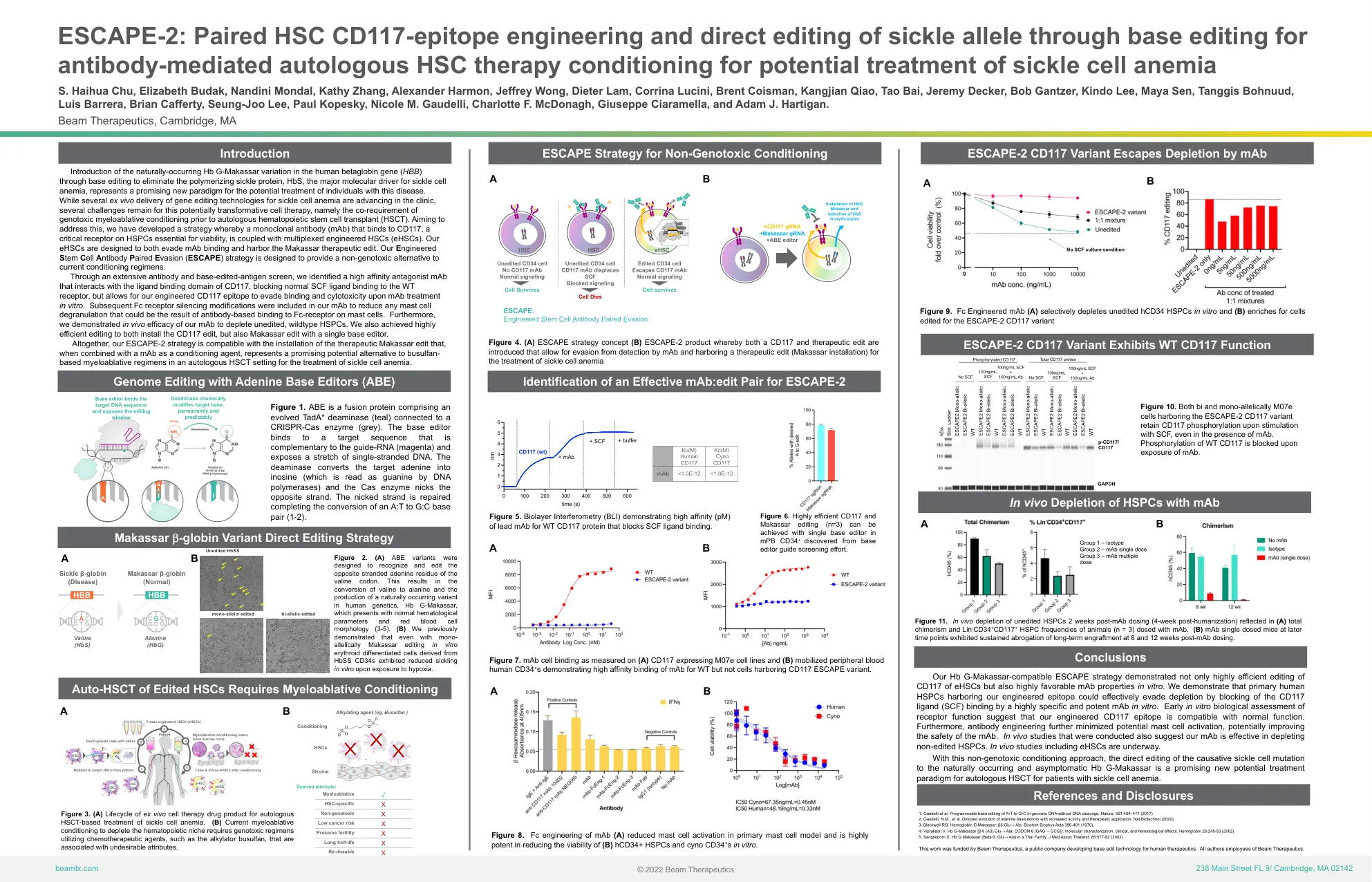
ESCAPE-2:配对 HSC CD117 表位工程和通过碱基编辑直接编辑镰状等位基因,用于抗体介导的自体 HSC 治疗 c
通过碱基编辑在人类β珠蛋白基因 ( HBB ) 中引入天然存在的 Hb G-Makassar 变异,以消除聚合镰状蛋白 HbS(镰状细胞性贫血的主要分子驱动因素),这代表了治疗这种疾病患者的潜在新模式。虽然临床上正在推进几种用于治疗镰状细胞性贫血的体外基因编辑技术,但这种具有潜在变革性的细胞疗法仍然存在一些挑战,即在自体造血干细胞移植 (HSCT) 之前必须进行基因毒性骨髓清除性预处理。为了解决这个问题,我们开发了一种策略,即将一种与 CD117 结合的单克隆抗体 (mAb) 与多重工程化 HSC (eHSC) 结合,CD117 是 HSPC 上对生存至关重要的关键受体。我们的 eHSC 旨在逃避 mAb 结合并携带 Makassar 治疗性编辑。我们的工程干细胞抗体配对逃避(ESCAPE)策略旨在为当前的预处理方案提供一种非基因毒性的替代方案。

X连锁严重联合免疫缺陷症的基因治疗
SCID-X1 背景和标准治疗结果 严重联合免疫缺陷病 (SCID) 是一组遗传异质性疾病,其特征是 T 淋巴细胞功能严重缺失,导致细胞和体液免疫力缺乏。X 连锁形式 (SCID-X1) 占病例的 30-40%,是由 IL2RG 基因编码的常见细胞因子受体 γ 链 (γ c) 缺陷引起的。γ c 最初被确定为高亲和力白细胞介素 2 受体的组成部分,是 IL-4、-7、-9、-15 和 -21 细胞因子受体复合物 (1) 的重要组成部分 (图 1A)。SCID-X1 中的分子缺陷导致 T 细胞和自然杀伤细胞发育完全缺失,以及终末 B 细胞成熟和功能缺陷。如果不接受治疗,由于复发性和机会性感染,预后均为致命的。异基因造血干细胞移植 (allo-HSCT) 可治愈该疾病,但对某些患者而言,结果仍不理想。(2) 原发性免疫缺陷治疗联盟 (PIDTC) 发表了两项关于异基因造血干细胞移植结果的回顾性研究,一项研究针对 240 名典型 SCID 患者,另一项更大规模的近期研究针对 662 名典型、渗漏性或 Omenn SCID 患者。(2) 两项研究均证实并完善了对生存至关重要的因素,例如存在活动性感染、使用匹配兄弟姐妹以外的捐赠者以及移植年龄。后一项研究首次报告了特定于某些遗传类型的 SCID 的结果,表明 SCID-X1 患者的生存率更高。SCID-X1 缺乏功能性 T 细胞和 NK 细胞,这是许多患者无需调节即可产生 T 细胞移植物的原因。供体衍生的 HSC 和祖细胞 (HSPC) 播种胸腺,并可能维持 T 细胞生成多年。然而,如果不使用骨髓抑制性调节剂,T 细胞和 NK 细胞以外的细胞大部分仍来自宿主。因此,未在移植前接受调节的 SCID-X1 患者不太可能不依赖免疫球蛋白替代或对疫苗接种产生反应 (2)。最近的研究清楚地表明,调节可促进更彻底的免疫重建,特别是使用烷化剂白消安,该剂可通过药代动力学进行调整,以实现受控的药物暴露并限制毒性。由于缺乏适合所有患者的匹配供体,以及急性和慢性移植物抗宿主病 (GVHD) 的并发症,标准异基因造血干细胞移植的成功率仍然有限。即使在这个年轻的年龄组中,急性和慢性 GVHD 的发病率也分别约为 20-25% 和高达 16% (2)。此外,使用不匹配的供体,特别是单倍体相合相关供体,与需要第二次异基因造血干细胞移植的风险更大相关 (2)。用表达正常 SCID-X1 致病基因的整合逆转录病毒载体转导自体 HSPC 细胞,将无需寻找匹配的供体,并消除 GVHD 和移植物排斥的风险。使用自灭活载体可提高 SCID-X1 基因治疗的安全性。表 1 显示了过去和当前 SCID-X1 基因治疗试验的摘要。巴黎和伦敦针对高风险患者进行了开创性的临床试验,希望实现强大的免疫重建,而没有异基因 HSCT 固有的 GVHD 风险。从骨髓中纯化 CD34+ 细胞,并用表达 IL2RG 转基因 (MFG-γ c) 的载体转导。总体而言,20 名男孩接受了基因治疗,其中 18 名患者 T 细胞重建迅速。在 17 名幸存者中,所有人通常都没有 SCID 相关感染。(3, 4)

CD34+ 造血干细胞和祖细胞体外编辑后 CRISPR 脱靶发现工具的比较分析
虽然存在多种研究 CRISPR 脱靶 (OT) 编辑的方法,但在临床相关编辑过程后,很少有方法在原代细胞中进行过头对头比较。因此,我们在体外造血干细胞和祖细胞 (HSPC) 编辑后比较了计算机模拟工具 (COSMID、CCTop 和 Cas-OFFinder) 和经验方法 (CHANGE-Seq、CIRCLE-Seq、DISCOVER-Seq、GUIDE-Seq 和 SITE-Seq)。我们使用 11 种与 Cas9 蛋白复合的不同 gRNA(高保真 [HiFi] 或野生型版本)进行编辑,然后对通过计算机模拟和经验方法确定的指定 OT 位点进行靶向下一代测序。我们平均每个向导 RNA (gRNA) 识别出少于一个 OT 位点,使用 HiFi Cas9 和 20-nt gRNA 生成的所有 OT 位点都可通过除 SITE-seq 之外的所有 OT 检测方法识别。这导致大多数 OT 提名工具具有高灵敏度,并且 COSMID、DISCOVER-Seq 和 GUIDE-Seq 获得了最高的阳性预测值 (PPV)。我们发现经验方法无法识别生物信息学方法未识别的 OT 位点。这项研究支持可以开发出既能保持高灵敏度又能保持 PPV 的精细生物信息学算法,从而能够更有效地识别潜在的 OT 位点,而不会影响对任何给定 gRNA 的彻底检查。

造血干细胞中临床相关的基因编辑治疗丙酮酸激酶缺乏症
丙酮酸激酶降低(PKD)是一种常染色体衰竭,是慢性非细胞性溶血性贫血的主要原因。pKD是由丙酮酸激酶,肝脏和红细胞(PKLR)基因中的突变引起的,该基因编码为红酮丙酮酸激酶蛋白(RPK)编码。rpk与红细胞(RBC)厌氧糖酵解的最后一步有关,负责维持正常的红细胞ATP水平。PKD的唯一治疗方法是同种异性造血茎和祖细胞(HSPC)移植,与显着的发病率和死亡率相关,尤其是PKD患者。在这里,我们通过PKLR内源性基因座的精确基因编辑来解决PKD的校正,以保持呈红生酶期间RPK酶的严格调节。我们合并了CRISPR-CAS9系统和供体的腺相关载体(RAAV)递送,以建立一个有效,安全且临床上适用的系统,以在人类造血祖先中RPK同工型的翻译起始位点敲击治疗序列。编辑的人类造血祖细胞在原发性和继发性免疫型小鼠中有效地重构的人伴有人伴有。源自编辑的PKD-HSPC的红细胞细胞恢复了正常的ATP水平,表明基因编辑后PKD红细胞生成中RPK功能的恢复。 我们的基因编辑策略可能代表了PKD患者RBC中RPK功能的终生疗法。红细胞细胞恢复了正常的ATP水平,表明基因编辑后PKD红细胞生成中RPK功能的恢复。我们的基因编辑策略可能代表了PKD患者RBC中RPK功能的终生疗法。

呼吸护理行动计划:2021-2026
更新以下通知于2021年5月7日添加。呼吸护理行动计划无意取代任何当前的临床指导,应用作与呼吸系统有关的健康和社会护理服务的总体战略。将在2021年夏季启动实施计划,以与呼吸社区合作推出本文档中概述的承诺。在本计划的第1章中,提到呼吸肺活量法作为气溶胶生成程序(AGP)。作为NSS国家IPC手册正在进行的文献评论的一部分,National Arhai进行了每月快速审查,以确保手动内容提供最新的证据。这些评论的输出为英国范围的IPC政策提供了信息,并为现有AGP列表的决定提供了由英国新和新兴的呼吸病毒威胁咨询小组(NERVTAG)决定的决定,并确定是否需要包括其他程序。查看全国一致的AGP清单。对可用文献的回顾表明,肺活量测定法不是定义为AGP。这意味着健康委员会和HSPC可以使用其当地感染的预防和控制团队来使用苏格兰Covid-19附录中包含的IPC指南重新引入肺活量测定法。查看社区IPC COVID-19附录。

人类造血干细胞中β-珠蛋白基因校正的发展是镰状细胞病的潜在耐用治疗
镰状细胞疾病(SCD)是最常见的严重单基因疾病,每年在全球范围内有300,000个出生。SCD是一种常染色体隐性疾病,是由-珠蛋白基因的第六个点突变(HBB)引起的。ex vivo -Globin基因校正在自体患者衍生的造血干细胞和祖细胞(HSPC)中可能有可能为SCD提供治疗性治疗。我们以前开发了一种CRISPR-CAS9基因靶向策略,该策略使用具有化学改良的指南RNA预处理的高保真性CAS9诱导重组腺相关病毒血清型6(RAAV6) - 介导的HBB基因校正HSPCS中的SCD引起的突变。在这里,我们证明了来自健康和SCD患者供体(GCHBB-SCD)的Plerixafor-Mobilized CD34 +细胞中HBB基因校正的临床前可行性和毒理学。我们在临床规模的GCHBB-SCD制造中最多可实现60%的HBB等位基因校正。移植到免疫缺陷型NSG小鼠中后,通过多核植入实现20%的基因校正。长期安全性,肿瘤性和毒理学研究表明,没有来自植入的GCHBB-SCD药物的造血,遗传毒性或肿瘤性异常的证据。一起,这些临床前数据支持该基因校正策略的安全性,功效和再现性,以启动SCD患者的1/2期临床试验。

基因组编辑模拟和逆转与骨髓增生性肿瘤相关的普遍突变
骨髓增生性肿瘤 (MPN) 会导致血细胞(如红细胞增多症)或血小板(原发性血小板增多症)的过度生成。JAK2 V617F 是许多 MPN 中最常见的体细胞突变,但之前在小鼠中对这种突变的建模依赖于转基因过度表达,并导致不同的表型,在某些情况下,这些表型归因于表达水平。CRISPR-Cas9 工程通过精确修改原代细胞中的内源性位点,为建模和潜在治愈遗传编码疾病提供了新的可能性。我们在此开发了“无疤痕”的 Cas9 试剂,用于在永生化人类红系祖细胞 (HUDEP-2)、CD34+ 成人人类造血干细胞和祖细胞 (HSPC) 以及免疫表型长期造血干细胞 (LT-HSC) 中创建和逆转 JAK2 V617F 突变。我们发现与内源性 JAK2 V617F 等位基因相关的体外增殖没有明显增加,但与野生型细胞共培养揭示了突变提供的竞争性生长优势。即使在没有造血细胞因子信号传导的情况下,获得 V617F 等位基因也会促进红系祖细胞的终末分化。综上所述,这些数据与 MPN 的逐渐进展的表现相一致,并表明与转基因过表达模型相比,内源性获得性 JAK2 V617F 突变可能产生更细微的表型。

Casgevy,INN-exagamglogene autotemcel(CD34+ 细胞)
该药品需要接受额外监测。这将可以快速识别新的安全信息。请医疗保健专业人员报告任何疑似不良反应。有关如何报告不良反应,请参见 4.8 节。 1. 药品名称 Casgevy 4 - 13 × 10 6 细胞/mL 输液分散液 2. 定性和定量组成 2.1 一般描述 Casgevy(exagamglogene autotemcel)是一种经过基因改造的自体 CD34 + 细胞富集群,含有通过 CRISPR/Cas9 在 BCL11A 基因的红细胞特异性增强子区体外编辑的造血干细胞和祖细胞 (HSPC)。 2.2 定性和定量组成 每个患者专用的 Casgevy 小瓶均含有 exagamglogene autotemcel,其浓度取决于批次,是经过基因改造的自体 CD34 + 细胞富集群。该药品包装在一个或多个小瓶中,总共含有 4-13 × 10 6 个细胞/毫升的富含 CD34 + 细胞的活细胞群,悬浮在冷冻保存溶液中。每瓶含有 1.5 至 20 毫升的输注分散液。药品的定量信息,包括要给药的小瓶数量(见第 6 节),均在运输所用冷冻运输器盖子内的批次信息表 (LIS) 中显示。已知作用的辅料 此药品每毫升含 50 毫克二甲基亚砜 (DMSO)。此药品每毫升含 3.5 毫克钠。有关辅料的完整列表,见第 6.1 节。3. 药物形式 输注分散液。半透明的细胞分散液,不含异物。

RPS19 编辑的 Diamond-Blackfan 贫血模型...
简介戴蒙德-布莱克凡贫血 (DBA) 是一种罕见的先天性骨髓衰竭疾病,通常在婴儿期表现为大细胞性贫血和红细胞减少症 (1, 2)。DBA 与腭裂、肾脏和心脏缺陷、生长迟缓等身体异常以及某些癌症风险增加有关 (3, 4)。虽然发育不全性贫血是儿童的主要特征,但老年患者也可能出现骨髓细胞减少、全血细胞减少和免疫缺陷,表明造血干细胞 (HSC) 受损 (5, 6)。经典的 DBA 是由 20 个小亚基或大亚基核糖体蛋白 (RP) 基因中的 1 个发生种系杂合功能丧失突变引起的,导致核糖体的生物合成和/或功能缺陷。较不常见的是,GATA1 (7)、EPO (8)、ADA2 (9) 和 TSR2 (10) 的突变会导致 DBA 样增生性贫血。最常见的 DBA 基因是 RPS19,大约 25% 的患者检测到突变。接下来最常见的突变基因是 RPL5 (~7%)、RPS26 (~7%) 和 RPL11 (~5%) (1)。目前对 DBA 的治疗方法包括铁螯合慢性红细胞输注;糖皮质激素(可促进红系祖细胞扩增)和异基因造血干细胞移植 (HSCT),所有这些疗法都与严重毒性有关。DBA 相关红系衰竭的机制尚不完全清楚。对患者造血干细胞和祖细胞 (HSPC) 的分析显示,红系祖细胞扩增存在缺陷,并伴有红系祖细胞病理性凋亡 (1, 11–14)。可能的解释包括整体翻译受损 (15, 16);BAG1 (17)、CSDE1 (17) 和 GATA1 (18, 19) 等红细胞生成所必需的转录本的选择性翻译受损;由于