XiaoMi-AI文件搜索系统
World File Search SystemMAD7

在Cybathlon锦标赛锦标赛上竞争残疾运动员:长期运动图像脑部计算机界面训练四边形CYB
我们测试了MAD7表达质粒的共转染和GRNA表达质粒作为RNP方案的替代方案。质粒转染具有不需要先前产生和纯化MAD7蛋白的优势。我们使用与RNP实验相同的GRNA序列靶向相同的基因。如图2所示,基于质粒的协议也可以生成indels,但显示的Indels水平低于RNP协议。类似于RNP协议,尽管不太明显,但4XNLS版本优于1XNLS版本。

MAD7:IP友好CRISPR酶
细胞,并重悬于裂解中(20 mM Tris,500 mM NaCl和10 mM Imidazole,pH 8.0),并补充了“完整的蛋白酶抑制剂鸡尾酒”(Merck,CAT,CAT#11697498001)。重悬于重悬酶后,苯并酶核酸酶(默克,CAT#E1014-5KU,每40 ml裂解物10μl)和溶菌酶(默克,CAT#10837059001,1 mg/ml裂解物),并加入冰上30分钟。细胞在Avestin乳液C-5均质器(15-20 kpsi)上被破坏,并通过离心(15,000 g,4°C,15分钟)去除不溶性细胞碎片。在10°C下进行所有随后的色谱步骤。清除的裂解物被加载在5 ml Histrap FF柱上(Cytiva,Cat#17525501)。将树脂用10列的洗涤液(裂解效果)洗涤(但使用20 mM咪唑)洗涤,并用10列的洗脱体洗脱蛋白(裂解效果,但用250毫米咪唑)洗脱。馏分合并,并在透析中稀释至25 mL(250 mM KCl,20 mM HEPES和1 mM DTT和1 mM EDTT和1 mM EDTA,pH 8.0)。使用透析膜管透析在10°C下透析,使用分子量切割为6-8 kDa(光谱/POR标准级再生纤维素,宽23毫米)的透析膜管。1-2小时后,将透析液替换,透析继续过夜。第二天,将透析样品在10 mM HEPE(pH 8)中稀释了两倍(pH 8),并立即加载在5 mL HITRAP肝素HP柱(Cytiva,CAT,CAT#17040601)上,并用Bu效率a(20 mM Hepes,150 mm Hepes,150 mm KCl,pH 8.0)。关注的分数被汇总并上升。将树脂用2柱体积洗涤,并使用bu虫B(20 mM Hepes,2m kcl,pH 8.0)的线性梯度在12柱体积上洗脱蛋白质。含量为2 mL;通过在120 mL SuperDex200凝胶滤光管(Cytiva#28989335)上注射上浓缩样品,以50 mM磷酸钠,300 mM NaCl,300 mM NaCl,0.1 mm EDTA,pH 7.5 AS分离bu e e e e e e e e e e o 进行了最终的色谱步骤。进行了最终的色谱步骤。

MAD7:IP友好CRISPR酶
环境自然光的非线性光学处理对于计算11成像和传感非常需要。在弱宽带不一致的12光下,强烈的光学非线性响应对于此目的至关重要。通过将2D透明光晶体管(TPTS)与13个液晶(LC)调节器合并,我们创建了一个光电子神经元阵列,该阵列允许在空间上构建宽度材料的较大的非线性对比度,从而使空间不相反的光振幅调制为空间不相互不到的光。我们制造了一个10,000像素阵列的光电神经元,17,并在实验上展示了一个智能成像系统,该系统可以立即启用18个输入眩光,同时保留了手机摄像头捕获的较弱的强度对象。19,这种智能眩光减少对于各种成像应用非常重要,包括20个自动驾驶,机器视觉和安全摄像机。不一致的宽带光的快速非线性处理21也可能在光学计算中找到应用,其中高度寻求环境光条件的22个非线性激活函数。23

使 ipsc 衍生细胞疗法的设计成为可能...
并表征重组 MAD7 以用于我们 iPSC 基因编辑平台中的核糖核蛋白 (RNP)。我们的工艺产生的蛋白质在配制后在溶液中是均质和单体的。此外,通过生物物理和功能表征测量,蛋白质稳定性在 -8080 C 下保持 6 个月。重组产生的 MAD7 的活性在 iPSC 中多个基因座的敲除 (KO) 和同源定向修复 (HDR) 效率方面与 Cpf1 相当。我们已经生成并测试了针对基因组中不同位点的多个 gRNA,并证明 MAD7 不会引起任何结构异常,这通过正交遗传表征测定确定。数据表明,重组 MAD7 CRISPR 核酸酶可以有效表达、纯化和配制,从而能够将哺乳动物细胞稳健而精确地改造为核糖核蛋白 (RNP)。我们目前正在使用我们的 MAD7 优化工艺来生成 MAD7 RNP,以便对具有多个基因编辑的治疗性 iPSC 衍生的 NK 和 T 细胞候选产品进行基因工程改造。Hunter Hoffman、Jill M. Carton、Buddha Gurung、Justin Bianchini、Shelby Keating、Michael F. Naso、Luis Borges
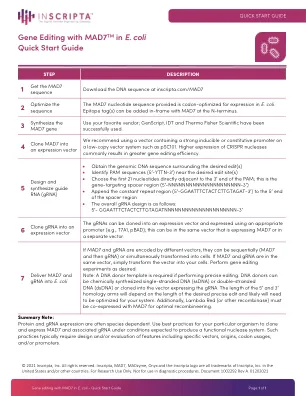
使用 MAD7TM 在大肠杆菌中进行基因编辑的快速入门指南
如果 MAD7 和 gRNA 由不同的载体编码,则可以依次(MAD7 然后是 gRNA)或同时将其转化为细胞。如果 MAD7 和 gRNA 在同一个载体中,只需将载体转化为细胞即可。根据需要进行基因编辑实验。注意:如果进行精确编辑,则需要 DNA 供体模板。DNA 供体可以是化学合成的单链 DNA (ssDNA) 或双链 DNA (dsDNA),也可以克隆到表达 gRNA 的载体中。5' 和 3' 同源臂的长度取决于所需精确编辑的长度,可能需要针对您的系统进行优化。此外,Lambda Red(或其他重组酶)必须与 MAD7 共同表达才能实现最佳重组。

ErCas12a CRISPR-MAD7 用于人类细胞、小鼠和大鼠的模型生成
摘要 MAD7 是从直肠真杆菌中分离出来的一种工程化的 2 类 VA 型 CRISPR-Cas (Cas12a/Cpf1) 系统。与 Cas9 类似,它是一种 RNA 引导的核酸酶,在大肠杆菌和酵母细胞中具有基因编辑活性。本文报告称,MAD7 能够分别在人类 HCT116 和 U2OS 癌细胞系中产生内源基因的插入/缺失和荧光基因标记。此外,MAD7 非常擅长在小鼠和大鼠胚胎中产生插入/缺失、小 DNA 插入(23 个碱基)和 1 至 14 kb 大小的较大整合,从而产生活产转基因动物。由于不同的原间隔区相邻基序要求、小引导 RNA 和高效的靶向基因破坏和插入,MAD7 可以扩展 CRISPR 工具箱,用于跨不同系统和模型生物进行基因组工程。


一个快速高效地设计多编辑工程的平台...
图 1:(A) Notch 的多重基因编辑平台使用属于 2 类 VA 型 CRISPR-Cas 家族的 MAD7 核酸酶,该核酸酶可识别富含胸腺嘧啶的 PAM ′YTTV′ 并产生双链交错断裂。(B) Notch 的符合 GMP 标准的 iPSC 系使用专有编辑协议针对临床相关基因进行批量编辑效率。我们的高通量 gRNA 筛选工作流程结合了通过 Synthego 的 CRISPR 编辑干扰 (ICE) 工具进行的可行性评估和插入缺失检测,然后通过靶向扩增子测序进行深入分析(左)。原代 T 细胞中敲除的表型验证(右)(C)与其他多重方法相比,我们的多重编辑方法实现了显着更高的编辑效率(左图)和显着降低的靶向易位率(中图)

CRISPR-MAD7 和 CRISPR-Cas9 在 Komagataella phaffii 中基因破坏的比较
摘要:基于 CRISPR(成簇的规律间隔的短回文重复序列)的技术是用于定点基因组修饰的强大、可编程工具。在成功改造并有效使用 CRISPR-Cas9 进行甲基营养酵母 Komagataella phaffii 的基因组工程后,人们希望有更多可用的核酸内切酶来增加实验灵活性,并在由于第三方的知识产权 (IPR) 而对工业研究有特定法律限制的情况下提供替代方案。MAD7 是一种工程化的 2 类 V 型 Cas 核酸酶,被推广为学术和工业研究的免版税替代品,由 Inscripta(美国加利福尼亚州普莱森顿)开发。本研究首次将CRISPR-MAD7用于K. phaffii基因组编辑,对编码甘油激酶1(GUT1)、红色荧光蛋白(DsRed)和zeocin抗性基因(Sh ble)的三个靶基因均获得了较高的基因编辑率(高达90%)。此外,还通过靶向K. phaffii中的259个激酶基因,系统地比较了CRISPR-MAD7和CRISPR-Cas9系统的基因组编辑效率。在这次大范围的测试中,与应用的CRISPR-MAD7工具箱(约23%)相比,CRISPR-Cas9具有更高的基因组编辑率,约为65%。

基因编辑技术的研究进度
1。摘要2。简介3。经典基因编辑工具4。精确编辑技术的开发4.1单基础编辑器4.2 Prime Editor 4.3双基本编辑技术5。转座子类编辑工具5.1集成5.2铸造6。开发新基因编辑工具6.1 SGN(结构引导的内切酶)6.2 MAD7 6.3 CRISPR-CASφ6.4 SPG和SPRY 7。的挑战和未来观点7.1不同的基因编辑工具存在很高的脱离目标问题,例如提高编辑效率7.2如何实现转座基因编辑工具的应用,例如在动物和工厂中进行整合和铸造7.3如何扩大编辑工具的编辑工具7.4如何启动编辑效率7.5启动eDing of new of ewnely eDITER 7. 5 eN.7 eNEDEN 7.是否能够启动eDing ofer ewnely eDITER,是否可以启动eDing eDITER的copecyme,以启动编辑工具,是否可以启动编辑工具,以启动编辑工具,是否可以启动编辑工具,是否可以启动编辑工具,是否可以启动编辑工具。各个国家逐渐在完全科学监督的前提下逐渐发布基因编辑的应用,以提高国民生活的质量8。作者贡献9。道德批准并同意参加10。确认11。资金12。利益冲突13。参考