XiaoMi-AI文件搜索系统
World File Search SystemMyc

通过抑制 ATPase RUVBL1/2 靶向胰腺癌中的 MYC 效应功能
摘要 目的 标志性致癌基因 MYC 驱动大多数肿瘤的进展,但小分子药物直接抑制 MYC 尚未进入临床试验。MYC 是一种依赖几种结合伙伴发挥作用的转录因子。因此,我们探索了通过胰腺导管腺癌 (PDAC) 中的相互作用组靶向 MYC 的可能性。 设计 为了在所有 MYC 结合伙伴中找出最合适的靶点,我们构建了一个靶向 shRNA 文库,并在培养的 PDAC 细胞和小鼠肿瘤中进行筛选。 结果 出乎意料的是,发现许多 MYC 结合伙伴对培养的 PDAC 细胞很重要,但在体内却不是必需的。然而,有些对自然环境中的肿瘤也是必不可少的,其中 ATPases RUVBL1 和 RUVBL2 排名第一。生长素-降解元系统降解 RUVBL1 导致培养的 PDAC 细胞停滞(而非未转化细胞),并导致小鼠的肿瘤完全消退,而此前免疫细胞浸润。从机制上讲,RUVBL1 是 MYC 建立致癌和免疫逃避基因表达所必需的,从而确定 RUVBL1/2 复合物是 MYC 驱动癌症中可用药的弱点。结论我们研究的一个含义是 PDAC 细胞依赖性受环境的强烈影响,因此应在体外和体内进行基因筛选。此外,生长素-降解元系统可应用于 PDAC 模型,从而允许在活体小鼠中进行靶标验证。最后,通过揭示 RUVBL1/2 复合物的核功能,我们的研究提出了一种使胰腺癌可能对免疫疗法敏感的药物策略。

氧化应激增强呼吸抑制剂对 MYC 驱动淋巴瘤的治疗作用
MYC 是多种肿瘤类型中的关键致癌驱动因素,但同时也使癌细胞具有一系列脆弱性,为有针对性的药物干预提供了机会。例如,抑制线粒体呼吸的药物会选择性地杀死 MYC 过表达的细胞。在这里,我们揭示了这种合成致死相互作用的机制基础,并利用它来提高呼吸复合物 I 抑制剂 IACS- 010759 的抗癌作用。在 B 淋巴细胞系中,异位 MYC 活性和 IACS- 010759 治疗加在一起会诱导氧化应激,从而导致还原谷胱甘肽的消耗和氧化还原稳态的致命破坏。这种效果可以通过抑制通过戊糖磷酸途径产生的 NADPH 或抗坏血酸(维生素 C)来增强,已知抗坏血酸在高剂量时可充当促氧化剂。在这些情况下,抗坏血酸与 IACS- 010759 协同作用,在体外杀死 MYC 过度表达细胞,并增强其对人类 B 细胞淋巴瘤异种移植的治疗作用。因此,复合物 I 抑制剂和高剂量抗坏血酸可能会改善高级别淋巴瘤和其他可能由 MYC 驱动的癌症患者的预后。

MYC 通过 microRNA 17/20a 调节 CSF1 表达,从而调节骨肉瘤中的肿瘤相关巨噬细胞
引言骨肉瘤 (OS) 是儿童和青少年中最常见且转移性最高的原发性骨肿瘤 (1)。尽管存在广泛的基因组畸变,但 OS 并没有特征性的 DNA 易位或可靶向的突变 (2)。因此,目前尚无针对 OS 的有效分子靶向疗法。然而,许多 OS 患者存在基因定义的体细胞 DNA 拷贝数改变,例如 8q24 染色体增加,约 20% 的 OS 患者有此表现 (3, 4)。8q24 基因座含有已知的致癌基因 c-MYC (MYC),它直接调节几种对不同细胞功能很重要的蛋白质编码和非编码基因,包括细胞周期调控、蛋白质生物合成、代谢、信号转导、转录和翻译 (5, 6)。已发现 MYC 在超过一半的人类癌症中失调 (7)。 8q24 区域扩增和 MYC 过度表达见于高级别癌前病变和侵袭性肿瘤,并且与不同人类肿瘤类型(包括 OS)的不良预后有关 (8–12)。除了对内在肿瘤细胞生物学的影响之外,MYC 的过度活化还会导致多种癌症的肿瘤免疫微环境 (TME) 发生改变 (13–15)。巨噬细胞是实体瘤(包括 OS)TME 中大量存在的细胞,通过释放独特的生长因子、细胞因子、趋化因子和酶 (16, 17),在宿主防御、组织修复、凋亡和组织稳态中发挥多功能作用。在成年人中,巨噬细胞在细胞因子巨噬细胞集落刺激因子 1 (M-CSF 或 CSF1) 的帮助下从外周血单核细胞分化。 CSF1 不仅调节单核细胞向巨噬细胞的分化,还通过与其受体 (CSF1R) 相互作用支持单核细胞/巨噬细胞的存活和增殖以及巨噬细胞的运动 (18)。肿瘤内致癌 MYC 在巨噬细胞调节中的作用已被证实。

增强的TP53重新激活破坏了MYC转录程序,并克服了急性髓样白血病中的Venetoclax耐药性
肿瘤抑制剂TP53经常在癌症中以突变的方式灭活,并通过抑制其阴性调节剂来重新激活。我们在这里cotarget MDM2和核出口XPO1至p53的最大转录活性。MDM2/XPO1抑制积累了核p53,并引起其转录靶标25至60倍。TP53调节MYC,MDM2/XPO1抑制作用破坏了C- MYC调节的转录组,从而导致急性髓样白血病(AML)的凋亡的协同诱导。出乎意料的是,耐Venetoclax的AML表达高水平的C-MYC,并且容易受到MDM2/ XPO1抑制体内的抑制作用。然而,MDM2/XPO1抑制后持续存在的AML细胞表现出静止和应激反应 - 相关表型。venetoclax克服了这种抗性,如单细胞质量旋转术所示。MDM2,XPO1和BCl2的三重抑制作用非常有效,对抗Venetoclax的AML体内。我们的结果提出了一种新型的,高度可翻译的治疗方法,利用p53重新激活以过度反应,反应适应压力的静脉抗体耐药性。

增强的TP53重新激活破坏了MYC转录程序,并克服了急性髓样白血病中的Venetoclax耐药性
肿瘤抑制剂TP53经常在癌症中以突变的方式灭活,并通过抑制其阴性调节剂来重新激活。我们在这里cotarget MDM2和核出口XPO1至p53的最大转录活性。MDM2/XPO1抑制积累了核p53,并引起其转录靶标25至60倍。TP53调节MYC,MDM2/XPO1抑制作用破坏了C- MYC调节的转录组,从而导致急性髓样白血病(AML)的凋亡的协同诱导。出乎意料的是,耐Venetoclax的AML表达高水平的C-MYC,并且容易受到MDM2/ XPO1抑制体内的抑制作用。然而,MDM2/XPO1抑制后持续存在的AML细胞表现出静止和应激反应 - 相关表型。venetoclax克服了这种抗性,如单细胞质量旋转术所示。MDM2,XPO1和BCl2的三重抑制作用非常有效,对抗Venetoclax的AML体内。我们的结果提出了一种新型的,高度可翻译的治疗方法,利用p53重新激活以过度反应,反应适应压力的静脉抗体耐药性。

针对脂肪酸结合蛋白会破坏多发性骨髓瘤细胞周期进程和 MYC 信号传导
摘要 多发性骨髓瘤是一种无法治愈的浆细胞恶性肿瘤,5 年生存率仅为 53%。迫切需要找到新的多发性骨髓瘤弱点和治疗途径。在此,我们确定并探索了一个新的多发性骨髓瘤靶点:脂肪酸结合蛋白 (FABP) 家族。在我们的工作中,用 FABP 抑制剂 (BMS3094013 和 SBFI- 26) 治疗骨髓瘤细胞,并在体内和体外检查细胞周期状态、增殖、细胞凋亡、线粒体膜电位、细胞代谢(耗氧率和脂肪酸氧化)和 DNA 甲基化特性。还使用 RNA 测序 (RNA-Seq) 和蛋白质组学分析评估了骨髓瘤细胞对 BMS309403、SBFI-26 或两者的反应,并通过蛋白质印迹和 qRT-PCR 确认。使用癌症依赖性图 (DepMap) 评估骨髓瘤细胞对 FABP 的依赖性。最后,挖掘了 MM 患者数据集 (CoMMpass 和 GEO) 中 FABP 表达与临床结果的相关性。我们发现,用 FABPi 或 FABP5 敲除 (通过 CRISPR/Cas9 编辑生成) 处理的骨髓瘤细胞在体外表现出增殖减少、凋亡增加和代谢变化。FABPi 在两种临床前 MM 小鼠模型中的体内结果好坏参半,这表明在临床应用之前需要优化体内递送、剂量或 FABP 抑制剂的类型。FABPi 在体外对线粒体呼吸产生负面影响,并降低 MM 细胞中 MYC 和其他关键信号通路的表达。临床数据显示,肿瘤细胞中 FABP5 表达高的患者总体生存率和无进展生存率较差。总体而言,这项研究将 FABP 家族确立为多发性骨髓瘤的潜在新靶点。在 MM 细胞中,FABP 具有多种作用和细胞作用,从而支持骨髓瘤进展。有必要对 MM 中的 FABP 家族进行进一步研究,尤其是对体内靶向这些家族的有效转化。

MYC驱动的线粒体DNA副本编号...
利益冲突:ESA是Janssen,Astellas,Sanofi,Dendreon,Pfizer,Amgen,Lilly,Lilly,Lilly,Lilly,Bayer,Astrazeneca,Bristol-Myers Myers Squibb,Clovis和Merck的有偿顾问/顾问;他从Janssen,Johnson&Johnson,Sanofi,Dendreon,Genentech,Novartis,Tokai,Bristol Myers-Squibb,Astrazeneca,Clovis和Merck获得了研究资金;他是AR-V7生物标志物技术的共同文化(专利编号CA2959336A)已获得许可获得Qiagen。SRD已从阿斯特拉斯(Astellas)向他的机构获得了研究资金。AMDM是默克和Cepheid的有偿顾问/顾问,并获得了Janssen和Myriad的研究资金。Sy从Bristol-Myers Squibb和Celgene,Janssen和Cepheid获得了他的机构研究资金,并曾担任Cepheid的顾问。他拥有Brahm Astra Therapeutics和Digital Harmonic的创始人权益。MCM是Clovis和Exelixis的付费顾问。

靶向MYC与表观遗传调节剂结合使用,在MLLR白血病中诱导协同的抗白血病作用,并同时提高免疫力
mll重排(MLL R)白血病与预后不良和对常规疗法的反应有限有关。此外,化学疗法会导致严重的侧面影响,并严重受到免疫系统的损害。因此,必须识别新型治疗策略。最近,我们通过使用簇状的定期插入的短篇小学重复序列(CRISPR)/cas9在CD34 +细胞中诱导CD34 +细胞中的染色体重排,开发了人类MLL RR白血病模型。该MLL R模型的真实性模仿患者白血病细胞,可用作新型治疗策略的平台。我们模型的 RNA测序揭示了MYC是促进造成发生的最重要的关键驱动因素之一。 然而,在临床试验中,BRD4抑制剂JQ-1导致间接阻断MYC途径仅显示适度的活动。 我们和其他人以前报道说,靶向MAT2A或PRMT5的表观遗传药物促进了MLL R细胞中的细胞死亡。 因此,我们将这些药物与JQ-1结合使用,从而增强了抗白血病效应。 更重要的是,我们发现T,NK和INKT细胞的激活,免疫调节细胞因子的释放以及抑制剂治疗后PD-1/PD-L1轴的下调导致细胞毒性提高。 总而言之,MYC和MAT2A或PRMT5的抑制作用驱动了MLL RL白血病的强大协同抗白血病活性。 此外,在组合抑制剂治疗后同时激活免疫系统,从而进一步提高了治疗效率。RNA测序揭示了MYC是促进造成发生的最重要的关键驱动因素之一。然而,在临床试验中,BRD4抑制剂JQ-1导致间接阻断MYC途径仅显示适度的活动。我们和其他人以前报道说,靶向MAT2A或PRMT5的表观遗传药物促进了MLL R细胞中的细胞死亡。因此,我们将这些药物与JQ-1结合使用,从而增强了抗白血病效应。更重要的是,我们发现T,NK和INKT细胞的激活,免疫调节细胞因子的释放以及抑制剂治疗后PD-1/PD-L1轴的下调导致细胞毒性提高。总而言之,MYC和MAT2A或PRMT5的抑制作用驱动了MLL RL白血病的强大协同抗白血病活性。此外,在组合抑制剂治疗后同时激活免疫系统,从而进一步提高了治疗效率。

用自体移植治疗的大型B细胞淋巴瘤患者MYC/BCL2表达和起源细胞的临床相关性
在常规化疗后,MYC和BCL2蛋白(双表达淋巴瘤[DEL])以及原产细胞(COO)的双重表达是弥漫性大B细胞淋巴瘤(DLBCL)患者的重要预后因素。我们研究了DEL和COO对用自体干细胞移植(ASCT)治疗的复发DLBCL患者的预后影响。鉴定出三百三名三个患者的组织样品。 在267名患者中取得了成功:161例(60%)是DEL/非双打(DHL),98(37%)为非DEL/非DHL,而8(3%)为DEL/DHL。 与非DEL/非DHL相比,DEL/DHL的总生存率较差,而DEL/非DHL的总生存率没有显着差异。 在多变量分析,DEL/DHL,年龄> 60岁和> 2个先前的疗法(但不是COO)上,是整体生存的重要预后因素。 当我们探索COO和的相互作用时鉴定出三百三名三个患者的组织样品。在267名患者中取得了成功:161例(60%)是DEL/非双打(DHL),98(37%)为非DEL/非DHL,而8(3%)为DEL/DHL。与非DEL/非DHL相比,DEL/DHL的总生存率较差,而DEL/非DHL的总生存率没有显着差异。在多变量分析,DEL/DHL,年龄> 60岁和> 2个先前的疗法(但不是COO)上,是整体生存的重要预后因素。当我们探索COO和
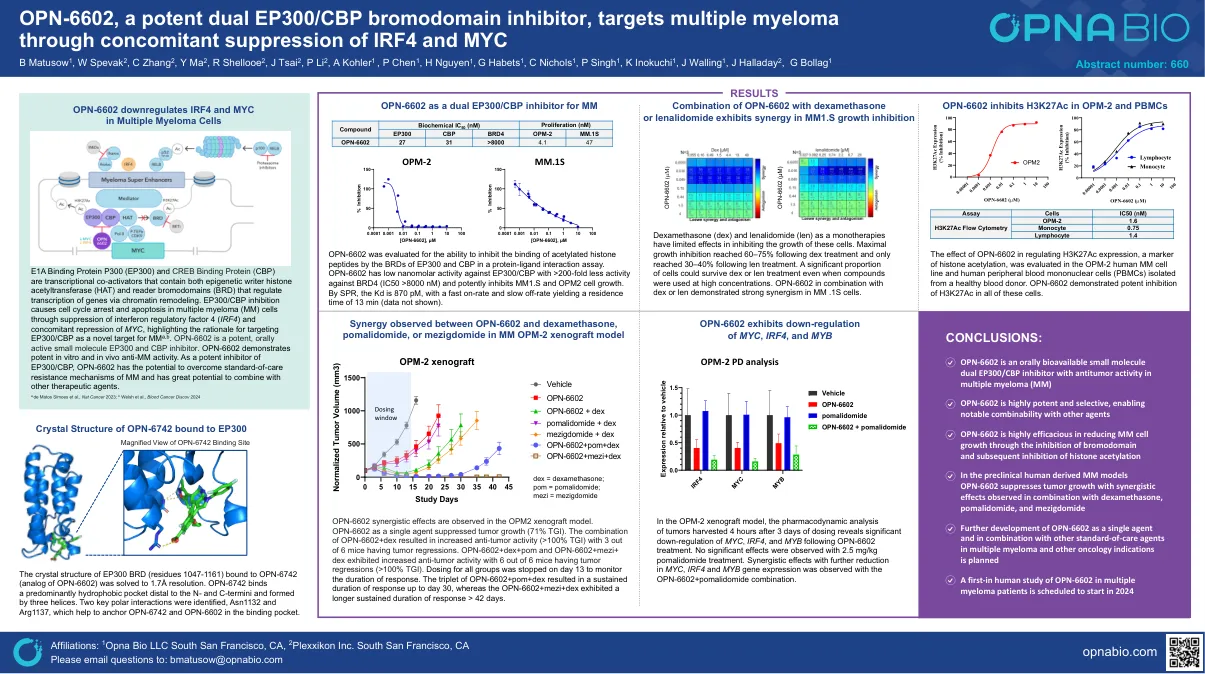
OPN-6602,一种有效的双EP300/CBP溴化域抑制剂,通过同时抑制IRF4和MYC
e1a结合蛋白p300(EP300)和CREB结合蛋白(CBP)是转录共激活因子,既包含表观遗传作者组蛋白乙酰基转移酶(HAT)和读取子溴化酶(BRD),并且通过染色质重塑调节基因的转录。EP300/CBP抑制作用通过抑制干扰素调节因子4(IRF4)和伴随抑制MYC导致多发性骨髓瘤(MM)细胞的细胞周期停滞和凋亡,并强调将EP300/CBP作为MM A,b,b,b。OPN-6602是一种有效的,有效的小分子EP300和CBP抑制剂。OPN-6602表现出体外和体内抗MM活性的有效性。 作为EP300/CBP的有效抑制剂,OPN-6602具有克服MM的护理标准抗性机制的潜力,并且具有与其他治疗剂相结合的巨大潜力。OPN-6602表现出体外和体内抗MM活性的有效性。作为EP300/CBP的有效抑制剂,OPN-6602具有克服MM的护理标准抗性机制的潜力,并且具有与其他治疗剂相结合的巨大潜力。