XiaoMi-AI文件搜索系统
World File Search SystemPairwise

从印度移民和印度 - 加拿大人中的非西方化向西方化肠道微生物组的过渡与饮食适应性有关
alpha多样性 - 使用16S扩增子序列,向前消失,反向读数在251个基础上被截断。框表示第一四分位数和第三四分位数之间的四分位间范围(IQR),水平线表示中位数,晶须是IQR的1.5倍以内的上和下值。alpha多样性。(a)Pielou的均匀度显示出显着差异(H = 85.7,P = 1.07E-17)。成对比较表明,印第安人的均匀均高于欧洲加拿大人(H = 56.2,Q = 6.51E-13),欧洲移民(H = 17.0,Q = 7.32e-05)和印度 - 加拿大人(H = 10.8,Q = 10.69e-03)。欧洲 - 加拿大的控制也比印度移民(H = 41.4,Q = 6.09e-10)和印度加拿大人(H = 21.7,Q = 8.10E-06)高得多。(b)香农的多样性显示出显着差异(H = 79.8,p = 1.89e-16)。成对比较表明,印第安人的多样性明显低于欧洲加拿大人(H = 56.7,Q = 3.91e-12),欧洲移民(H = 32.9,Q = 4.94E-08),印度 - 加拿大人,H = 14.3,Q = 14.3,Q = 2.59e-04)和Indo-Indo-Indo-Mignmants(H = 6. H = 17)6.66,6.66。欧洲 - 加拿大对照组的得分也明显高于印度移民(h = 20.0,q = 2.62e-05)和印度 - 加拿大人(h = 14.4,q = 2.59e-04)。beta多样性 - 使用shot弹枪序列,向前的反复读取和反向读数以10个碱基对修剪,固定最大长度为120个碱基对。使用Bray Curtis差异和加权Unifrac探索了Beta多样性,并使用成对的置换式多元方差分析(PermanOva)来测试组之间的差异。(c)Bray Curtis原理坐标分析(PCOA)图显示,在前两个轴上捕获了45.3%的变异。与Bonferroni校正的成对比较显示,大多数群体之间存在显着差异,印第安人和西方同类群的印第安人和印度移民的明显聚类。(d)加权unifrac PCOA图显示,在前两个轴上捕获了61.8%的变化,还检测到显着差异。成对比较结果可以在附录E中找到。印度的肠道表现出了杆菌和蛋白质细菌的占主导地位,欧洲加拿大人表现出很高的富公司。印度移民的消失(挥发性和/或与人类工业化社会有效)相关的大量分类单元,包括Eubacterium和Erysipelotrichaceae。印度 - 加拿大人拥有诸如Collinsella等消失的分类单元,以及Blossum(Bloom或在城市化/现代化社会中被选中)的大量丰富,例如Anaerostipes和Blautia。
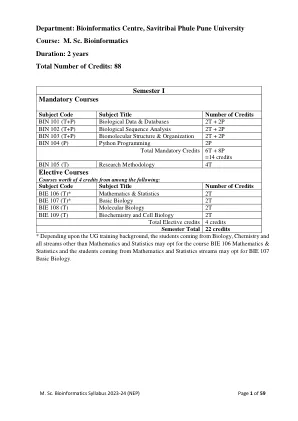
Savitribai Phule Pune University M.Sc. II物理化学
questions Syllabus: Theory Biomolecular sequence analysis (2) o Overview o Concepts Analysis of single sequence (2) o Nucleotide o Protein Pairwise sequence alignment algorithms (3) o Needleman & Wunsch o Smith & Waterman Scoring matrices for Protein and Nucleotide sequences (3) o MDM/PAM series o BLOSUM series o CSW 数据库相似性搜索(6)o快速o爆炸多个序列比对算法(6)o clustalw o肌肉o dalign o dalign o t-coffee序列徽标,共识和模式(3)序列配置文件的基本概念,配置文件的衍生;应用程序(5)O Gribskov的配置分析方法o Psi-Blast实践/教程:目标:本课程将使学生能够:了解如何使用各种算法进行生物分子序列分析了解各种参数在相应算法中使用各种参数及其对结果的影响学习和练习编码和练习一些差异,以<练习<<<<

美国尾流湍流重新分类的发展(特邀)
本文介绍了背景信息,并提供了联邦航空管理局 (FAA) 尾流湍流计划 RECAT(即重新分类)特定方面的状态更新。RECAT 的基本前提是,可以使用更完整的尾流相关参数集来改进尾流分离,而不是使用基于最大起飞重量的现有 FAA Order JO 7110.65 分类尾流湍流分离最小值。然后,此过程可以安全地降低尾流湍流分离最小值,使其低于 FAA Order JO 7110.65 中规定的最小值。本文介绍了 RECAT 的整体三阶段方法,最终目标是实现动态成对分离。目前,第二阶段或基于静态成对的尾流湍流分离已准备好由联邦航空管理局实施。本文介绍了分析方法,包括 RECAT 第二阶段开发中使用的数据源和严重程度指标。

Photobiology.pdf
在分类学多样性中,主要数据是税收 - 原子树,这些树被转化为物种之间成对距离。在功能多样性中,主要数据是物种特征,这些特征转移到物种之间的成对距离,然后转移到种类特征的树木中。使用树木的论点是,这样一个偏差物种将具有很小的影响,因为它仅评估一次,而不是评估其与所有其他物种的距离(Petchey and Gaston,2006年)。函数TREEDIVE实现功能分化定义为连接所有物种的性状树状图中的总分支长度,但不包括树的不必要的根部段(Petchey和Gaston,2002,2006)。该示例使用上一章的分类距离。这些首先转换为层次结构(实际上是它们的原始形式,然后在将它们转换为距离之前)

innovastemcell台式11月24日。cdr
促销细胞表皮球菌ATCC的生长14990 AER 6小时的culɵaaɵooon营养琼脂II培养基(对照),并在补充5%金属息座的培养基上。意味着不共享上标的Leter是显着不同的(Tukey成对比较,P值≥0.05)。

混合会员随机块模型
考虑由成对测量组成的数据,例如对象对之间是否存在链接。例如,这些数据出现在蛋白质相互作用和基因调控网络、作者-收件人电子邮件集合和社交网络的分析中。使用概率模型分析成对测量需要特殊的假设,因为通常的独立性或可交换性假设不再成立。在这里,我们引入了一类用于成对测量的方差分配模型:混合成员随机块模型。这些模型结合了实例化密集连接块(块模型)的全局参数和实例化连接中节点特定变异性的局部参数(混合成员)。我们开发了一种用于快速近似后验推理的通用变分推理算法。我们展示了混合成员随机块模型的优势,并将其应用于社交网络和蛋白质相互作用网络。关键词:分层贝叶斯、潜在变量、均值场近似、统计网络分析、社交网络、蛋白质相互作用网络

单纯霍普菲尔德网络
Hopfield 网络是一种人工神经网络,它通过选择循环连接权重和更新规则将记忆模式存储在神经元的状态中,使得网络的能量景观在记忆周围形成吸引子。我们可以在这种使用 N 个神经元的网络中存储多少个稳定、足够吸引人的记忆模式?答案取决于权重和更新规则的选择。受生物学中集合连通性的启发,我们通过添加集合连接并将这些连接嵌入到单纯复形中来扩展 Hopfield 网络。单纯复形是图的高维类似物,它自然地表示成对和成组关系的集合。我们表明,我们的单纯 Hopfield 网络增加了记忆存储容量。令人惊讶的是,即使连接仅限于与全成对网络大小相同的小随机子集,我们的网络仍然优于成对网络。这样的场景包括非平凡的单纯拓扑。我们还测试了类似的现代连续 Hopfield 网络,为改进 Transformer 模型中的注意力机制提供了一条潜在的有希望的途径。
GeneTargeter:在疟原虫寄生虫中的基因组编辑中自动化,恶性疟原虫
丰富度与更好的编辑活动有关[54,55]。均聚物据报道偶尔会降低SGRNA效率[54-56]。 可以用两种算法之一来计算sgrna裂解预期位点的预测概率的目标分数:(1)Doench等人开发的原始规则集2分数。 cas9 sgrnas [57],并以方位角更新(github.com/microsoftresearch/azimuth);或(2)用于与CAS12A SGRNA一起开发的Cindel分数[53]。 最后,可用的靶向活动评分算法包括HSU等人开发的分数。 [58]和Doench等人开发的切割确定(CFD)得分。 [57]。 两者都是基于选择的SGRNA与目标基因组中所有其他可能的SGRNA之间成对比较的分数,并且使用系数矩阵确定成对得分,该系数矩阵在SGRNA中考虑了不匹配位置,以及在CFD得分的情况下确定了不匹配的身份。 因为两个分数的系数矩阵均来自均聚物据报道偶尔会降低SGRNA效率[54-56]。可以用两种算法之一来计算sgrna裂解预期位点的预测概率的目标分数:(1)Doench等人开发的原始规则集2分数。cas9 sgrnas [57],并以方位角更新(github.com/microsoftresearch/azimuth);或(2)用于与CAS12A SGRNA一起开发的Cindel分数[53]。最后,可用的靶向活动评分算法包括HSU等人开发的分数。[58]和Doench等人开发的切割确定(CFD)得分。[57]。两者都是基于选择的SGRNA与目标基因组中所有其他可能的SGRNA之间成对比较的分数,并且使用系数矩阵确定成对得分,该系数矩阵在SGRNA中考虑了不匹配位置,以及在CFD得分的情况下确定了不匹配的身份。因为两个分数的系数矩阵均来自

使用结构图嵌入的快速蛋白质结构搜索
考虑到局部几何形状[5],坐标对齐[6]和3D Zernike的描述符[7,8],已经开发了多种方法来比较,对齐和搜索[1] [1] [1] [2,3,4]。由于蛋白质结构比序列[9]更保守[9],这些方法已被证明在远程同源性检测[10],蛋白质分类[11]中有用[11],从结构[12]推断功能[12],聚类大数据库[13,14]并评估结构预测的准确性。最高的精度方法倾向于根据DALI等坐标[3]进行仔细的比较,但是搜索大型结构数据库,例如Alphafold蛋白结构数据库[15,16]或ESM宏基因组图[17] [17]使用这些方法很慢。最近,foldseek [18]通过将一级序列转换为一系列学到的局部特长基序来解决了这个问题。然后,它使用生物信息学中快速序列搜索的丰富历史记录大大减少查询的成对比较时间与数据库的每个成员。为了进一步减少搜索时间,应更快地将成对比较步骤进行。

通过偏好学习和人体工程学增强了人类 - 肉体协作的框架
行业5.0旨在优先考虑人类运营商,专注于他们的福祉和能力,同时进行人类和机器人之间的合作,以提高效率和生产力。协作机器人的整合必须确保人类运营商的健康和福祉。的确,本文解决了以人类机器人协作(HRC)方案中基于偏好的优化算法提出以人体工程学评估来提高基于偏好的优化算法的必要性,以改善工作条件。HRC应用程序包括在对象处理任务期间优化协作机器人最终效果。以下方法(AMPL-RULA)利用了一种主动的多首选项学习(AMPL)算法,这是一种基于偏好的优化方法,在其中要求用户通过在几个候选人之间表达成对的偏好来迭代提供定性反馈。要解决身体健康,符合人体工程学的性能指数,快速上肢评估(RULA)与用户的成对偏好相结合,以便可以计算最佳设置。实验测试以验证该方法,涉及机器人执行的对象处理过程中的协作组装。结果表明,所提出的方法可以在简化协作任务时改善操作员的物理工作量。