机构名称:
¥ 1.0
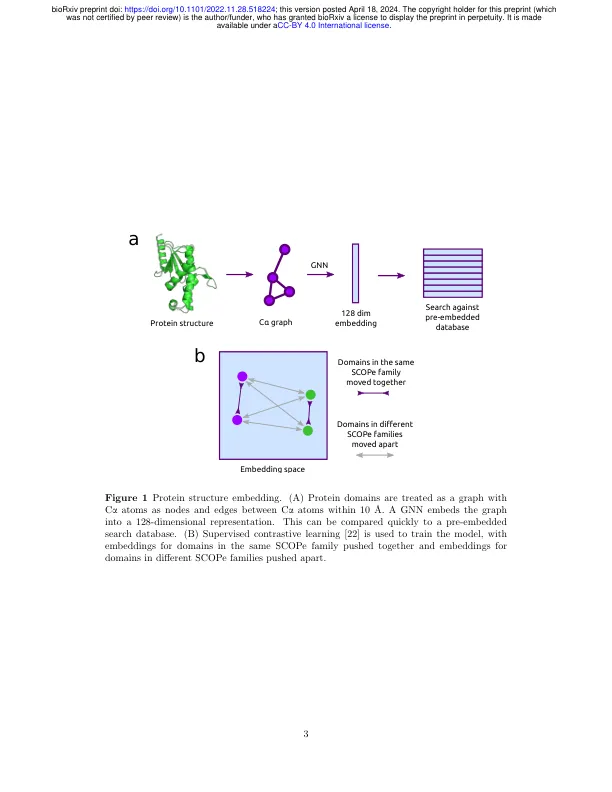
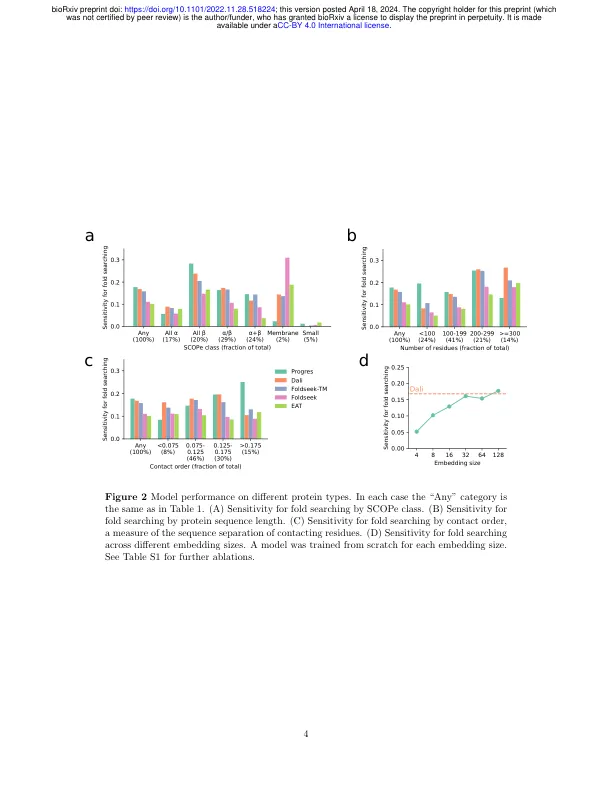
考虑到局部几何形状[5],坐标对齐[6]和3D Zernike的描述符[7,8],已经开发了多种方法来比较,对齐和搜索[1] [1] [1] [2,3,4]。由于蛋白质结构比序列[9]更保守[9],这些方法已被证明在远程同源性检测[10],蛋白质分类[11]中有用[11],从结构[12]推断功能[12],聚类大数据库[13,14]并评估结构预测的准确性。最高的精度方法倾向于根据DALI等坐标[3]进行仔细的比较,但是搜索大型结构数据库,例如Alphafold蛋白结构数据库[15,16]或ESM宏基因组图[17] [17]使用这些方法很慢。最近,foldseek [18]通过将一级序列转换为一系列学到的局部特长基序来解决了这个问题。然后,它使用生物信息学中快速序列搜索的丰富历史记录大大减少查询的成对比较时间与数据库的每个成员。为了进一步减少搜索时间,应更快地将成对比较步骤进行。
使用结构图嵌入的快速蛋白质结构搜索
主要关键词



相关文件推荐