XiaoMi-AI文件搜索系统
World File Search SystemPairwise

使用高阶的简单复杂传染
摘要 - 神经退行性疾病的特征是复杂的蛋白质错误折叠的大脑内传播。例如,当前的发现突出了2种特定错误折叠蛋白在阿尔茨海默氏症中的作用,这些蛋白质被认为使用脑纤维作为高速公路传播。先前的研究通过模拟模型或基于机器学习的预测变量调查了这种扩散,这些预测因素采用大脑连接组作为基础扩散网络。但是,构造的结构连接组仅描述图中节点之间的成对连接。高阶相互作用复杂网络比正常图提供了显着的优势,因为它们可以捕获超出简单的成对关系船的交互。蛋白质错误折叠和聚集通常涉及正常图的合作行为或群体动力学,其专注于单个边缘,无法充分代表。蛋白质错误折叠的非线性和多尺度可能更适合更丰富的高阶模型。在这项研究中,我们研究了高阶网络在这种情况下是否可以提供改进的拟合和解释能力。更具体地说,我们采用淀粉样蛋白β的简单复杂传染模型来预测蛋白质错误折叠的扩散。Simplicial Cronagion复合物在2年的地平线和其他结果中,阿尔茨海默氏症患者在所有大脑区域的预测蛋白质沉积中产生了0.030的平均重建误差,胜过先前的研究,尤其是对于错误折叠的蛋白质的病例稳定增长。尽管时间范围有限,但这项研究突出了结合先进网络分析以捕获跨神经网络蛋白质聚集的复杂动力学的潜力。临床相关性 - 这项研究突出了高阶网络在阿尔茨海默氏症中提高错误折叠蛋白传播的预测的潜力,从而更好地洞悉了蛋白质聚集动力学。

可编程通用光子处理器中的情境性、连贯性和维度的实验认证
高维状态的量子叠加使得加密协议中的计算速度和安全性都得以提升。然而,层析成像过程的指数复杂性使得这些属性的认证成为一项具有挑战性的任务。在这项工作中,我们使用由飞秒激光写入技术制造的六模通用光子处理器实现的成对重叠测量,通过实验认证了针对不断增加的维度的量子系统的相干性见证。特别是,我们展示了所提出的相干性和维度见证对于维度高达 5 的量子比特的有效性。我们还展示了量子询问任务中的优势,并表明它是由量子语境性推动的。我们的实验结果证明了这种方法对于可编程集成光子平台中量子属性认证的有效性。


可编程通用光子处理器中的上下文性、相干性和维度的实验认证
高维状态的量子叠加可以提高加密协议的计算速度和安全性。然而,层析成像过程的指数级复杂性使得这些属性的认证成为一项具有挑战性的任务。在这项工作中,我们使用由飞秒激光写入技术制造的六模通用光子处理器实现的成对重叠测量,通过实验认证了针对不断增加的维度的量子系统的相干性见证。特别是,我们展示了所提出的相干性和维度见证对于维度高达 5 的量子位的有效性。我们还展示了在量子询问任务中的优势,并表明它是由量子语境性推动的。我们的实验结果证明了这种方法对于可编程集成光子平台中量子属性认证的有效性。

lavaan:潜在变量分析
efa 函数本质上是 lavaan 函数的包装器。它生成模型语法(针对给定数量的因子),然后调用 lavaan() 将这些因子视为应旋转的单个块。该函数仅支持单个组。分类数据照常处理,首先计算适当的(例如四分法或多分法)相关矩阵,然后将其用作 EFA 的输入。还(有限地)支持两级数据。然后在内部和之间提取相同数量的因子。promax 旋转方法(取自 stats 包)仅为方便起见提供。因为 promax 是一个两步算法(首先是方差最大,然后是斜向旋转以获得简单结构),所以它不使用 gpa 或成对旋转算法,因此不提供标准误差。


MSC Defense
响应肾脏分配的紧迫挑战,其特征是对器官的需求不断增长,该研究旨在为该问题开发出数据驱动的解决方案,该解决方案也包含了利益相关者的价值观。这项研究的主要目的是使用“成对的肾脏在线调查”中的数据来学习与肾脏分配有关的个人和群体级别偏好的方法。通过评估指标,使用机器学习分类器进行了三个级别的两个不同的数据集评估两个级别 - 个人,组和稳定性。单个级别数据模型可以预测各个参与者的偏好,组级别数据模型汇总参与者的偏好,而稳定性级别数据模型(组扩展小组级别)评估了这些偏好随时间的稳定性。
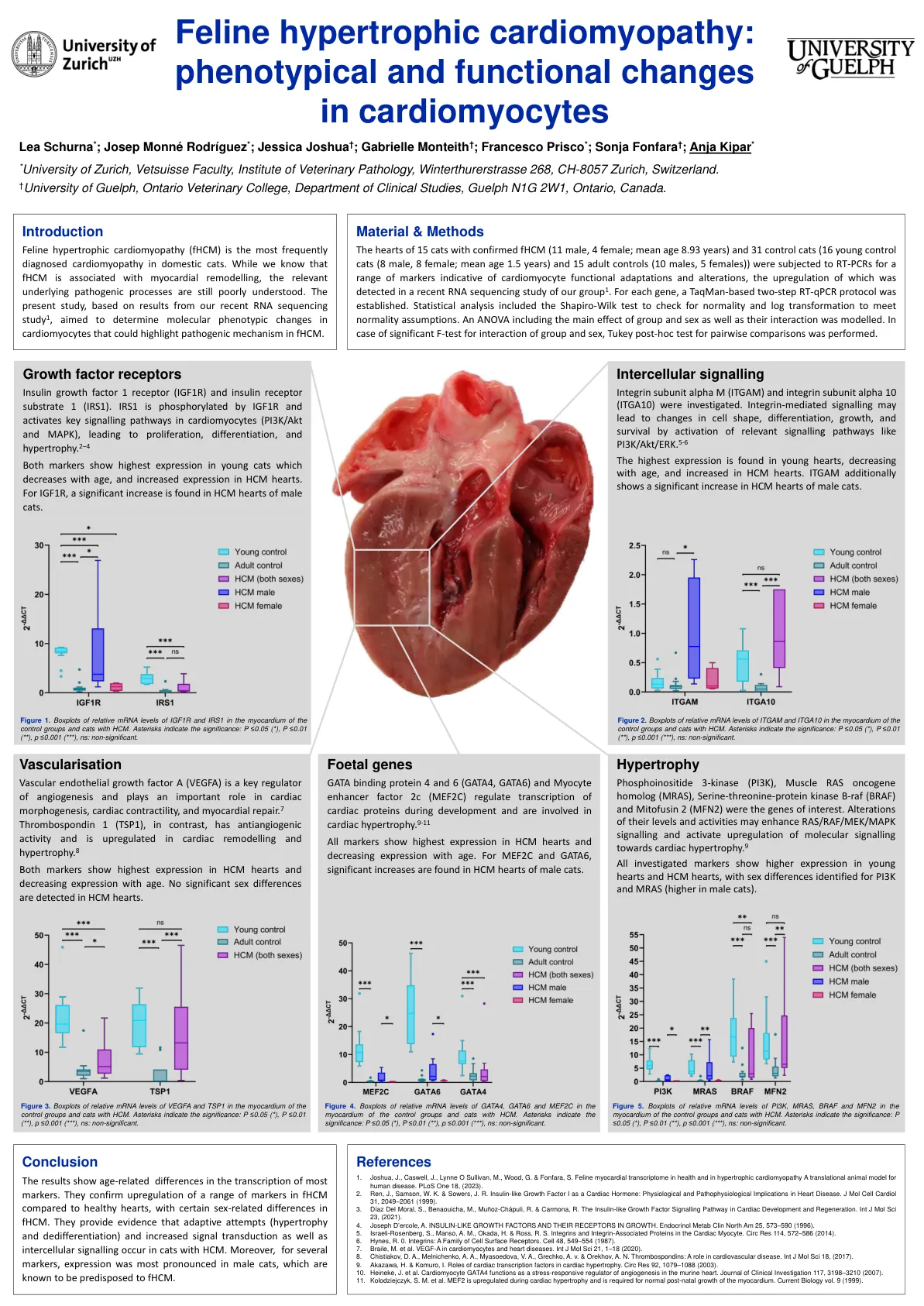
心肌细胞的表型和功能变化
对 15 只确诊为 fHCM 的猫(11 只雄性,4 只雌性;平均年龄 8.93 岁)和 31 只对照猫(16 只幼年对照猫(8 只雄性,8 只雌性;平均年龄 1.5 岁)和 15 只成年对照猫(10 只雄性,5 只雌性))的心脏进行 RT-PCR,检测一系列表明心肌细胞功能适应和改变的标志物,在我们最近对第 1 组的 RNA 测序研究中检测到了这些标志物的上调。对于每个基因,建立了基于 TaqMan 的两步 RT-qPCR 方案。统计分析包括 Shapiro-Wilk 检验以检查正态性,以及对数变换以满足正态性假设。建立了包括组别和性别的主效应及其相互作用的方差分析模型。如果组别和性别的相互作用的 F 检验显著,则进行 Tukey 事后检验以进行成对比较。

来自未培养微生物的新型微型 CRISPR–Cas13 系统可有效降解 SARS-CoV-2 序列和流感病毒
Prodigal28进行基因预测。使用BLASTP39对S蛋白进行两两比对,得到相似度>50%的BLASTP比对结果。然后使用MCL根据BLASTP结果对S蛋白进一步聚类,以创建S蛋白簇。我们从得到的S蛋白组中选择一个代表性基因组,共产生78个代表性基因组。接下来,我们使用滑动窗口法提取冠状病毒基因组中所有独特的30nt或22nt序列,分别为7,132,478和5,931,021个独特序列。独特序列按其出现的冠状病毒基因组数量降序排列。我们使用78个代表性基因组为每个独特序列创建条形码。独特序列

评估伪细菌中基因表达的载体及其在Aplysina Marine Sponge研究中的应用多蛋白质结构与flodmason
蛋白质结构是超出序列的保守,这使得多重结构比对(MSTA)对于分析远距离相关的蛋白质必不可少。计算预测方法已大大扩展了我们可用蛋白质结构的存储库,需要快速准确的MSTA方法。在这里,我们介绍了一种渐进式MSTA方法,该方法利用了成对结构对准器Foldseek的结构字母,用于多次对齐数十万个蛋白质结构。foldmason计算置信度得分,提供交互式可视化,并在准确的结构预测时代提供了大规模蛋白质结构分析的必要速度和准确性。使用flaviviridae糖蛋白,我们证明了Foldmason的MSTAS如何支持暮光区下方的系统发育分析。foldmason是免费的开源软件:foldmason.foldseek.com和web服务器:search.foldseek.com/foldmason。