XiaoMi-AI文件搜索系统
World File Search SystemRRNA

shot弹枪宏基因组学捕获的微生物多样性比有针对性的16S rRNA基因测序用于现场标本和保存的博物馆标本
尽管头部计算机断层扫描(CT)对轻度创伤性脑损伤(TBI)的患者的需求逐渐增加,但只有少数人患有颅内病变,需要神经外科干预。因此,本研究旨在评估机器学习(ML)技术在巴西帕拉纳州玛林加地区医院的轻度TBI患者筛查中的适用性。这是一项使用ML技术的观察性,描述性,横截面和回顾性研究,以开发一种方案,该方案预测哪些初始诊断为轻度TBI的患者应建议用于头部CT。在测试模型中,他线性极端梯度的提升是最佳算法,其灵敏度最高(0.70±0.06)。我们的预测模型可以帮助筛查温和的TBI患者,帮助卫生专业人员管理资源利用,并提高患者护理的质量和安全性。
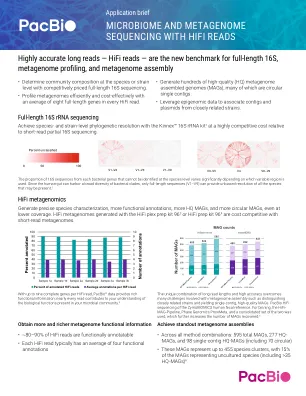
应用简介-使用 HiFi 进行宏基因组测序-...
1. 应用说明 – Kinnex 16S rRNA 试剂盒用于全长 16S 测序 2. Johnson, JS 等人 (2019) 评估 16S rRNA 基因测序在物种和菌株水平微生物组分析中的应用。《自然通讯》。10(1),5029。 3. 程序和清单 – 使用 HiFi plex 制备试剂盒 96 制备多重全基因组和扩增子文库 4. 程序和清单 – 使用 HiFi 制备试剂盒 96 制备全基因组文库 5. Gehrig, J. 等人 (2022) 找到合适的选择:评估短读和长读测序方法以最大限度提高临床微生物组数据的效用。《微生物基因组学》,8(3),10.1099/mgen.0.000794。 6. Portik, DM 等人(2024) 使用长读组装、分箱和合并方法从人类肠道微生物群中高度准确地组装宏基因组。bioRxiv。doi:https://doi.org/10.1101/2024.05.10.593587 7. 概述 – HiFi 应用选项和测序建议。8. 程序和清单 – 使用条形码引物扩增细菌全长 16S 基因。9. 程序和清单 – 从 16S rRNA 扩增子制备 Kinnex 文库
服务指南PACBIO REVIO 16S
所有样本中都包含分析,并且提供了RAW HIFI读取数据,以供客户更喜欢进行自己的分析。PACBIO HIFI全长16S数据使用QIIME2和DADA2进行质量过滤,并“将”“变形”到高质量扩增子单个变体(ASV)。ASV分类采用两种方法:针对基因组分类学数据库(GTDB R207)的共识一致性分类(使用VSEARZERCH)高一致性,以及使用三个数据库的基于贝叶斯的基于机器学习的分类(DADA2),使用三个数据库:GTDB R207,SILVA R207,SILVA RRNA DATABASE(v138)由核糖体数据库项目(RDP)补充,以更好地分类低充足的ASV。

符合欧洲药典第 2.6.7 章和 USP <77> 草案当前修订版本的新型支原体实时 RT-PCR 检测试剂盒
监管指南要求用于支原体污染检测的 PCR 试剂盒具有高灵敏度,而 DNA 靶标仅在生物体中以低拷贝水平存在,这种灵敏度呈上升趋势。这就是为什么传统的基于 DNA 的 PCR 在试图保持检测的稳健性和可靠性时逐渐达到极限的原因。实时逆转录 PCR 提供了一种克服此问题的智能解决方案。每个在 DNA 水平上可检测到的基因在目标生物体内也可作为转录本。特别是 16S rRNA 区域,一个高度保守的 rRNA 操纵子,是支原体检测的目标,在一个细胞内有多个 RNA 拷贝。RNA 水平上多个靶标的出现有助于用 PCR 检测较少数量的细胞。逆转录聚合酶使 RNA 拷贝可作为 cDNA 靶标,因此与基于 DNA 的基本 PCR 检测相比,可用的 PCR 靶标成倍增加。确实,这种方法无法对 PCR 结果进行任何定量解释,因为 16S rRNA 基因的 RNA 拷贝数非常灵活,但当涉及到需要“是”或“否”答案的质量控制问题时,定量输出不是必需的。这种方法特别简单,因为逆转录已经在 PCR 反应混合物中实施。

使用 QIIME 2 进行可重复、交互、可扩展和可扩展的微生物组数据科学
致编辑 — 过去 20 年里,DNA 测序和生物信息学技术的飞速发展大大提高了我们对微生物世界的了解。这种日益增长的了解涉及微生物的巨大多样性;微生物区系和微生物组如何影响疾病 1 和医学治疗 2;微生物如何影响地球的健康 3 ;以及微生物组生物技术在医学 4 、法医 5 、环境 6 和农业 7 应用的新兴探索。这方面的工作大部分是由标记基因调查(例如,细菌/古细菌的 16S rRNA 基因、真菌内部转录间隔区和真核生物的 18S rRNA 基因)推动的,这些调查以不同程度的分类特异性和系统发育信息来分析微生物区系。该领域目前正在转向整合其他数据类型,如代谢物 8 、宏蛋白质组 9 或宏转录组 9,10 图谱。

叙事综述了宏基因组方法的应用,以研究口咽微生物组和传染病之间的联系
不同16S rRNA可变区检测不同细菌群落的敏感性可能会改变微生物组研究中的比较。某些地区比其他地区提供了更好的识别(Bukin等,2019); for example, V1-V3 regions can discriminate between staphylococcal populations ( Conlan et al., 2012 ), whereas V3-V4 were shown to be better at discriminating a greater number of taxa in the vaginal microbiota such as Gardnerella vaginalis, Bifidobacterium bifidum , and Chlamydia trachomatis Graspeuntner et al.(2018)。16S基于RRNA的技术在处理过程中的高风险,测序误差以及解释不同操作分类单元的存在(OTUS)(OTUS)时也可能受到限制(Quince等,2009,2011; Youssef等,2009)。

拓扑复杂的 RNP 底物中的序列引导 RNA 重塑
摘要:DEAD-box ATPase 是 RNA 生物学各个方面必不可少的普遍存在的酶。然而,这些酶有限的体外催化活性与它们复杂的细胞作用不一致,最显著的是它们在核糖核蛋白 (RNP) 组装过程中驱动大规模 RNA 重塑步骤。我们描述了 60S 核糖体生物合成中间体的低温电子显微镜结构,揭示了 DEAD-box ATPase Spb4 的上下文特异性 RNA 解旋如何导致 rRNA 二级结构的广泛、序列定向重塑。多个顺式和反式相互作用稳定了催化后高能中间体,从而驱动 rRNA 结构域 IV 内根螺旋结构的组织。该机制解释了如何利用 DEAD-box ATPase 有限的链分离来提供非平衡方向性并确保高效准确的 RNP 组装。

假喹啉和N1-甲基丙啶作为RNA疗法和疫苗发育的有效核苷酸类似物
pseudouridine(c)位点。9–13细胞内C形成是由一种称为假喹啉合酶的酶催化的。14假喹啉合酶可以分为两个主要家族:较大蛋白质中的独立假酮合酶和假喹啉合酶结构域。独立的假性合酶包括在细菌和细菌和酵母中发现的trua中的TRUA。在真核生物中,发现了几个假喹啉合酶结构域。胞核H/ACA盒小核仁核糖蛋白(SNORNPS)具有dyskerin(CBF5)成分,可在rRNA,SNRNA和雌激素酶RNA中催化假硫苷化。nop10是H/ACA snornps的另一个组成部分,它参与了伪苷活性。14

评估和优化从附生植物学样品中提取微生物DNA的裂解方法
分析附生植物圈中的微生物群落可能具有挑战性,尤其是在应用基于测序的技术时,由于植物来源的生物分子(例如核酸)的干扰。对附生微生物组的最新研究的综述表明,机械和酶促方法都广泛使用。在这里,我们评估了两种裂解方法对DNA提取产率,纯度,完整性和微生物16S rRNA基因拷贝数在不同提取条件下的每种模板基因组DNA的影响。此外,使用16S rRNA基因扩增子测序研究了对细菌群落组成,多样性和可重复性的影响。酶促裂解方法产生的DNA增加了一到两个数量级,但DNA质量是次优的。相反,使用Me-Chanical方法制备的样品显示出高的DNA纯度,尽管产量较低。出乎意料的是,机械裂解显示出比酶裂解更高的DNA完整性数(DIN)。16S rRNA扩增子测序结果表明,通过机械破坏制备的样品表现出可重复的相似的微生物群落组成,无论提取条件如何。相比之下,酶促裂解方法在不同的提取条件下导致分类学组成不一致。这项研究表明,机械DNA破坏比酶促破坏更适合附生层样品。