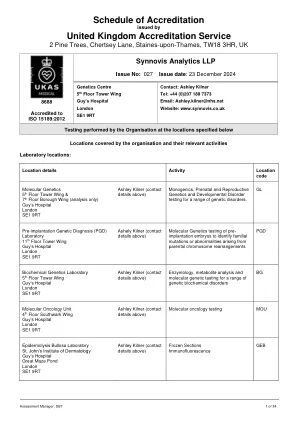
XiaoMi-AI文件搜索系统
World File Search SystemRYR1

脂质在冷冻EM条件下调节RYR1的开放概率
ryanodine受体(RYRS)是负责从肌质和内质网释放的细胞内四聚离子通道。在三种已知的哺乳动物RYR同工型中,RYR1对于肌肉收缩至关重要,并且已被广泛研究。RYRS的细胞质暴露多域碎片整合了多个细胞信号,这些信号调节通道门控和与Ryrs生理开放概率的小偏差导致危及生命的疾病。冷冻EM在揭示RYR门控机制的近原子细节方面发挥了作用,但在冷冻EM条件下RYR1的开放概率明显低于电生理研究中观察到的,这使RYR1门控模型的结构研究变得复杂。在这里,我们提出了一项冷冻EM研究,研究了在脂质浓度不同的CHAP中溶解的RyR1的开放概率。我们发现,将脂质浓度从0.001%增加到0.05%,将RYR1开放概率从16升至84%。但是,RYR1重组为脂质纳米盘仍关闭。我们在以最高脂质浓度重建的地图中建模了72个脂质分子。这些发现表明,脂质在冷冻EM条件下调节RYR1门控的关键作用,并提出了RYRR1门控调制的结构研究的最佳脂质模拟物。

RYR1 相关肌病的最新进展
&& ];然而,RYR1-RM 患者的临床和组织病理学特征与易患恶性高热和/或劳力性横纹肌溶解症的患者有相当大的重叠。这种重叠凸显了对 RYR1 突变患者进行警惕的临床管理以及考虑恶性高热风险的必要性 [11]。根据意大利的一项研究报告,研究人员调查了 153 名患有核心肌病(核心和微核心)的患者中的 RYR1 突变 [12]。其中,他们发现 68 例至少有一个 RYR1 基因突变。此外,他们还检查了核心肌病的基因型-表型相关性,发现孔域中的突变与胎儿运动减少、挛缩和足部畸形有关。研究还强调,RYR1 基因其他区域突变的患者表现出不同的临床表现,包括肌肉强度、呼吸功能的差异以及脊柱侧弯的存在,这表明 RYR1-RM 的复杂性和多样性 [12]。最近的研究结果扩大了 RYR1-RM 的病理谱。研究人员在两例管状聚集性肌病 (TAM) 患者中发现了 RYR1 突变 (p.Thr2206Met 和 p.Gly2434Arg),该病的特征是 CK 水平升高和由重复的肌肉收缩或暴露于寒冷引起的阵发性僵硬。值得注意的是,这两种突变也主要在恶性高热易感性 (MHS) 个体中检测到,突显了与 RYR1 突变相关的重叠临床表现 [6

RyR1 相关肌病的基因治疗
蛋白质 [ 1 , 3 ],导致细胞质钙稳态改变。许多不同的肌病与 RYR1 致病变异有关,例如中央核病 (CCD)、多小核病 (MmD)、中心核肌病 (CNM)、先天性纤维类型不平衡 (CFTD),现在这些肌病被称为“RyR1 相关肌病”或 RyR1-RM。RyR1-RM 的治疗受到 RYR1 基因和蛋白质的许多特性的限制,其中包括基因的大小(转录本为 15 kb)和蛋白质(超过 5,000 个氨基酸),形成超过 2 MDa 的同型四聚体。目前,人们正在探索两种治疗方案:使用化学分子的药物治疗和基因治疗,前者包括欧洲药品管理局和美国食品药品管理局分类的 DNA 或 RNA 导向治疗。两种治疗策略都有各自的特点,因此各有优缺点。一般而言,药物治疗通常使用小化学分子,定期(每天或每周)口服或静脉注射。药物治疗针对部分或全部下游病理生理机制。基因治疗通常使用大 DNA/RNA 分子,一次或多次给予患者。基因治疗直接针对不同病理生理机制上游的受影响基因或其产物,因此其作用涵盖了广泛的后果,理论上可以通过同一种治疗逆转所有这些后果。目前,药物疗法是 RyR1-RM 临床试验中唯一有效的治疗方法。最近完成了一项随机、双盲、安慰剂对照试验(I/II 期),研究对象为抗氧化剂治疗(N-乙酰半胱氨酸),但不幸的是,该治疗既没有降低之前发现的氧化应激升高,也没有显著改善患者的身体活动能力 [4]。正在进行的一项试验使用一种所谓的 Rycal 分子 (S48168) 来调节 RyR1 通道功能(ClinicalTrials.gov 标识符 NCT4141670,[5]),以减少由一组致病变异引起的钙漏。除了药物治疗外,基因治疗现在似乎也是这些遗传疾病的一种有吸引力的解决方案。事实上,使用药理学疗法很有吸引力,因为它很容易实施(例如当分子以口服形式提供时,如 NAC 或 S48168),在出现

RyR1 相关肌病的基因疗法 - CmyPath
蛋白质 [ 1 , 3 ],导致细胞质钙稳态改变。许多不同的肌病与 RYR1 致病变异有关,例如中央核病 (CCD)、多小核病 (MmD)、中心核肌病 (CNM)、先天性纤维类型不平衡 (CFTD),现在这些肌病被称为“RyR1 相关肌病”或 RyR1-RM。RyR1-RM 的治疗受到 RYR1 基因和蛋白质的许多特性的限制,其中包括基因的大小(转录本为 15 kb)和蛋白质(超过 5,000 个氨基酸),形成超过 2 MDa 的同型四聚体。目前,人们正在探索两种治疗方案:使用化学分子的药物治疗和基因治疗,前者包括欧洲药品管理局和美国食品药品管理局分类的 DNA 或 RNA 导向治疗。两种治疗策略都有各自的特点,因此各有优缺点。一般而言,药物治疗通常使用小化学分子,定期(每天或每周)口服或静脉注射。药物治疗针对部分或全部下游病理生理机制。基因治疗通常使用大 DNA/RNA 分子,一次或多次给予患者。基因治疗直接针对不同病理生理机制上游的受影响基因或其产物,因此其作用涵盖了广泛的后果,理论上可以通过同一种治疗逆转所有这些后果。目前,药物疗法是 RyR1-RM 临床试验中唯一有效的治疗方法。最近完成了一项随机、双盲、安慰剂对照试验(I/II 期),研究对象为抗氧化剂治疗(N-乙酰半胱氨酸),但不幸的是,该治疗既没有降低之前发现的氧化应激升高,也没有显著改善患者的身体活动能力 [4]。正在进行的一项试验使用一种所谓的 Rycal 分子 (S48168) 来调节 RyR1 通道功能(ClinicalTrials.gov 标识符 NCT4141670,[5]),以减少由一组致病变异引起的钙泄漏。除了药物治疗外,基因治疗现在似乎也是这些遗传疾病的一种有吸引力的解决方案。事实上,使用药理学是有吸引力的,因为它很容易实施(例如当分子以口服形式提供时,如 NAC 或 S48168),在出现

使用 HDAC 和 DNA 甲基转移酶抑制剂治疗先天性肌病小鼠模型可改善其肌肉强度
摘要 迄今为止,尚无针对先天性肌病患者的治疗方法,这种肌肉疾病会导致患者的生活质量低下。在约 30% 的病例中,先天性肌病患者携带瑞安诺丁受体 1 (RYR1) 基因的显性或隐性突变;隐性 RYR1 突变伴随着骨骼肌中 RyR1 表达和含量的降低,并与纤维营养不良和肌肉无力有关。重要的是,隐性 RYR1 突变患者的肌肉表现出 II 类组蛋白去乙酰化酶和 DNA 基因组甲基化的含量增加。我们最近创建了一个 p.Q1970fsX16+ p.A4329D RyR1 突变的小鼠模型,该突变与患有隐性 RyR1 相关多微核病的严重儿童携带的突变同源。 RyR1 突变小鼠的表型重现了携带隐性 RYR1 突变的患者的临床表现的许多方面。我们用两种靶向 DNA 甲基化酶和 II 类组蛋白脱乙酰酶的药物组合治疗复合杂合小鼠。在这里,我们表明,用靶向表观遗传酶的药物治疗突变小鼠可改善肌肉强度、RyR1 蛋白质含量和肌肉超微结构。这项研究为药物治疗与隐性 RYR1 突变相关的先天性肌病患者提供了概念证明。

8688医疗多重
Congenital myopathy / Congenital muscular dystrophy (COL6A1, COL6A2, COL6A3, COL12A1, FKRP, FKTN, LAMA2, LARGE1, POMGNT1, POMGNT2 (GTDC2), POMT1, POMT2, COL4A1, COL4A2, DAG1, DPM1, DPM2, DPM3, dolk, ISPD, GMPPB, b3galnt2, chkb, plec, sil1, b4gat1 (b3gnt1), pomk (sgk196), itga7, Them5, Micu1, act1, cfl2, dnm2, tbd1, mbt1, mbt1, mbt1, myh8,neb,ryr1,sepn1,tnni2,tnnt1,tnnt3,tpm2,tpm3,stim1,ecel1,cdc78,kbtbd10(kbtbd10),klhl40(kbtbd5)(kbtbd5),mybdbd),mybdbd),mybd) ),mybd),mybd),mybd) Lamp2, VMA21, STAC3, lmod3, MEGF10, epg5, ttn, adamts15, cacna1s, CNTN1, Doc7, Golga2, Hacd1 (PLPLA), inpp5k, klhl9, msto1, Mtm18, mybpp, mybpp, mybpp, mybpp, mybpp srpk3,them38a,trappc11)
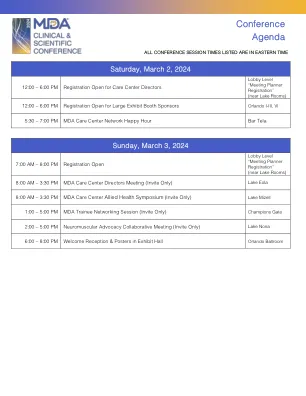
2024年3月2日,星期六
快速射击临床案例评论第2部分:肌肉疾病(CE认证)约翰·戴(John Day),医学博士,博士(联合主席)巴克里·埃尔希克(Bakri Elsheikh),MBBS,MBBS,FRCP,FAAN,FAAN(共同主席)成人肌动型性疾病性疾病性疾病性疾病性疾病症状疾病的妊娠1型类型1型fullenkamp,phd,PHD,phd诊断:先天纤维类型不成比例?Natalie Katz, MD, PhD Diagnosis Unknown: Suspected Pediatric Dystrophinopathy Natalie Katz, MD, PhD Titinopathy Diagnosis After Decades-Long Odyssey Jennifer Roggenbuck, MS, CGC Adult- Onset Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) Jennifer Roggenbuck, MS, CGC LGMD 2T/R19是由于GMPPB(肌病与NMJ赤字相结合)Matthew Wicklund,MD,Faan恶性高温,CK升高和RYRR1在GMPPB(肌病结合NMJ赤字)中引起的。
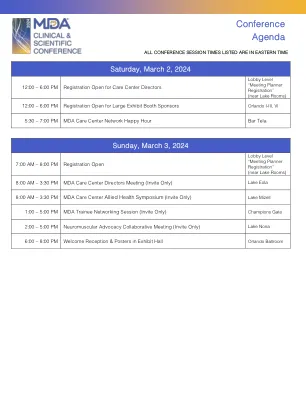
2024-MDA-Conference-Agenda-32。 ...
快速射击临床案例评论第2部分:肌肉疾病(CE认证)约翰·戴(John Day),医学博士,博士(联合主席)巴克里·埃尔希克(Bakri Elsheikh),MBBS,MBBS,FRCP,FAAN,FAAN(共同主席)成人肌动型性疾病性疾病性疾病性疾病性疾病症状疾病的妊娠1型类型1型fullenkamp,phd,PHD,phd诊断:先天纤维类型不成比例?Natalie Katz, MD, PhD Diagnosis Unknown: Suspected Pediatric Dystrophinopathy Natalie Katz, MD, PhD Titinopathy Diagnosis After Decades-Long Odyssey Jennifer Roggenbuck, MS, CGC Adult- Onset Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) Jennifer Roggenbuck, MS, CGC LGMD 2T/R19是由于GMPPB(肌病与NMJ赤字相结合)Matthew Wicklund,MD,Faan恶性高温,CK升高和RYRR1在GMPPB(肌病结合NMJ赤字)中引起的。

Rycarma Therapeutics宣布了新的领导力和公司名称,引入了
波士顿,马萨诸塞州 - 2025年1月7日 - Rycarma Therapeutics,Inc。,以前被称为Armgo Pharma(“公司”或“ Rycarma”),这是一家临床阶段的生物技术公司,开发了一流的小分子治疗方法,用于心血管疾病和骨骼肌肉疾病,该公司的领导力和公司的名字均已变化。致“ Rycarma Therapeutics,Inc。”并引入了心力衰竭的新计划。Rycarma这个名字引用了该公司在Ryanodine受体(RYR)生物学中的基础工作和领导力,并在建立Rycals®的治疗潜力时,这是一种新颖的小分子药物。Rycarma的新计划着重于开发其主要候选ARM210在心力衰竭中,弹射分数减少,这影响了全球数百万的人。ARM210目前也正在研究Ryanodine受体1-相关肌病(RYR1-RM),这是由RYR1基因突变引起的一组罕见的神经肌肉疾病。