XiaoMi-AI文件搜索系统
World File Search SystemSMAD

脂肪衍生的间充质干细胞治疗,用于反向博来霉素诱导的实验性肺纤维化
特发性肺纤维化(IPF)是一种慢性进行性呼吸道疾病。可以说,免疫细胞亚群之间的复杂相互作用,再加上对疾病病理生理学的不完全了解,阻碍了成功疗法的发展。尽管努力了解其病理生理学并开发有效的治疗方法,但IPF仍然是致命的疾病,需要探索新的治疗选择。间充质基质/干细胞(MSC)疗法在IPF的实验模型中表现出了希望,但是需要进一步研究以了解其治疗作用。这项研究旨在评估博来霉素诱导的肺纤维化模型中脂肪衍生的间充质干细胞的治疗作用。首先,从小鼠获得MSC细胞,并使用流式细胞仪和细胞分化培养方法进行表征。然后,将成年C57BL/6小鼠暴露于气管内滴注博来霉素,并在第14天与MSC进行逆转模型。在第14、21或28天评估实验组。此外,还用MSCS上清液或MSC处理了受TGF-β1挑战的肺成纤维细胞,以探索肺纤维化逆转的机制。间充质干细胞从小鼠脂肪组织中成功分离,并根据其分化能力和细胞表型进行表征。MSC或其上清液的存在刺激了肺纤维化细胞的增殖和迁移。MSC上清液减少了肺胶原蛋白沉积,提高了Ashcroft评分,并降低了与肺纤维化相关物质的基因和蛋白质表达。博来霉素挑战者的小鼠表现出严重的间隔增厚和突出的纤维化,MSC治疗有效地反转了。MSC上清液可以抑制TGF-β1/SMAD信号通路,上清液可促进成纤维细胞自噬。总而言之,这项研究表明,MSCS上清液治疗与MSC一样有效,可以恢复博来霉素诱导的肺纤维化的核心特征。当前的研究表明,MSC上清液减轻了体内BLM诱导的肺纤维化。在体外实验进一步表明,MSC上清液可以抑制TGF-β1/SMAD信号传导途径,以抑制TGF-β1诱导的成纤维细胞激活,并通过调节p62表达来促进成纤维细胞自噬。这些发现有助于越来越多的证据体系,支持MSC在IPF中的细胞治疗医学中的治疗应用。

名称:Peter McCaffery Center/单位:医学科学学院大学:Aberdeen Post标题:IMS神经生物学教授兼主管Web
名称:Peter McCaffery Center/单位:医学科学学院大学:Aberdeen帖子标题:IMS Neurobiology教授兼主管Web-Site:http://www.abdn.ac.uk/ims/staff/staff/details.php/details.php?id=p.j.j.mccaffery 1。研究输出 - 最近的一些相关出版物Bremner JD,Shearer KD,McCaffery p。视网膜酸和情感障碍:关联的证据。临床精神病学杂志(出版社,2011年)。Shearer KD,Goodman TH,Ross AW,Reilly L,Morgan PJ,McCaffery PJ。下丘脑中视黄酸信号传导的光周期调节。神经化学杂志112,246-57(2010)。Silvestri C,Narimatsu M,Von I,Liu Y,Tan NB,Izzi L,McCaffery PJ,Wrana JL,Attisano L.识别SMAD/FOXH1靶标的全基因组方法揭示了FOXH1在减虫酸调节和后代大脑中的关键作用。发育细胞14,411-23(2008)。Bremner JD,McCafferyP。情感疾病中视黄酸的神经生物学。神经药理学与生物精神病学的进展15,315-31(2008)

癌症幸存者动脉粥样硬化发展的可能的分子机制
癌症幸存者接受治疗面临的患有动脉粥样硬化心血管疾病(CVD)的风险增加,但潜在的机制仍然难以捉摸。最近的研究表明,化学疗法可以推动衰老癌细胞获得被称为衰老相关的干性(SAS)的增殖表型。这些SAS细胞表现出增长和对癌症治疗的耐药性,从而导致疾病进展。内皮细胞(EC)衰老与包括癌症幸存者在内的动脉粥样硬化和癌症有关。癌症治疗方式可以诱导EC衰老,从而导致SAS表型的发展和随后的癌症幸存者动脉粥样硬化。因此,针对显示SAS表型的衰老EC是一种治疗该人群动脉粥样硬化CVD的治疗方法的希望。本综述旨在提供对EC中SAS诱导及其对癌症幸存者动脉粥样硬化的贡献的机械理解。我们深入研究了EC衰老的基础机制,这些机制响应于流动的流量和电离辐射,这些辐射在动脉粥样硬化和癌症中起着关键作用。关键途径,包括P90RSK/TERF2IP,TGFβR1/SMAD和BH4信号传导作为癌症治疗的潜在靶标。

径向神经胶质通过整合素A V B 8 -TGF B 1信号促进小胶质细胞发育
小胶质细胞多样性来自固有的遗传程序与环境衍生的信号之间的相互作用,但是这些过程如何在发育中的大脑中展开和相互作用尚不清楚。在这里,我们表明在径向胶质祖细胞中表达的径向胶质胶质表达的整联蛋白β8(ITGB8)激活了小胶质细胞表达的TGF B 1,允许小胶质细胞发育。在这些祖细胞中,ITGB8的域限制缺失建立了互补的区域,其发育中被阻止的“变形障碍”小胶质细胞持续到成年。在没有自分泌TGF B 1信号传导的情况下,我们发现小胶质细胞采用了类似的畸形表型,从而导致神经运动症状与ITGB8突变小鼠几乎相同。相比之下,缺乏TGFβ信号传感器SMAD2和SMAD3的小胶质细胞具有较小的极化表型,相应地相应的严重神经运动功能障碍。最后,我们表明,非典型(独立于SMAD的)信号传导部分抑制了疾病和发育相关的基因表达,为在损伤或疾病背景下采用小胶质细胞发育信号通路提供了令人信服的证据。

Myostatin和心脏
摘要:Myostatin(生长分化因子8)是转化生长因子-β超家族的成员。它主要由骨骼肌分泌,尽管少量的肌蛋白素也由心肌和脂肪组织产生。肌抑制素与激活素IIB膜受体结合,以激活下游细胞内典型的SMAD2/SMAD3途径,并在非SMAD(非传统)途径上作用。对转基因动物的研究表明,肌抑制素的过表达减少心脏质量,而去除肌抑制素的作用相反。在这篇综述中,我们总结了该蛋白在与心脏有关的条件下的潜在诊断和预后价值。首先,在肌抑制素无小鼠中,左心室内径以及舒张压和收缩压量大于野生型小鼠的各个值。Myostatin可能被分泌为负反馈回路的一部分,该反馈循环降低了促进生长因子的释放和对肥大刺激的响应的能量重编程的影响。另一方面,人类和动物的数据都表明,肌抑制素参与了慢性心力衰竭过程中心脏恶病和心脏纤维的发展。对肌生抑制素在这种情况下的作用的理解可能会基于肌生信号抑制引发靶向疗法的发展。

TNIK抑制对肿瘤和相关免疫细胞具有双重协同作用
因子 β (TGF- β )/SMAD、磷脂酰肌醇 3-激酶 (PI3K)/AKT 和 RAS/RAF,所有这些都是潜在的治疗靶点。此外,Traf2 和 Nck 相互作用蛋白激酶 (TNIK) 最近被确定为 β -catenin 和 T 细胞因子 4 (TCF-4) 转录复合物的调节成分。针对这种蛋白质的几种小分子化合物(NCB0846(NCB)、甲苯咪唑(MBZ)等)已显示具有抗肿瘤作用。[5] 据报道,在黑色素瘤中,肿瘤内在的活性β-catenin信号传导导致T细胞排斥和对抗PD-L1 /抗CTLA-4单克隆抗体疗法的耐药性。[7] 据报道,TNIK抑制剂(TNIKi)可以增强急性感染中T细胞向效应细胞的分化。[6] 由于对肿瘤和免疫细胞可能有不同的影响,TNIKi的体内效应很复杂,而且目前尚无全面的了解它如何影响CRC微环境。我们最近开发了一种多重免疫表型分析方法(FAST),可以通过细针抽吸(FNA)对肿瘤微环境中的细胞进行连续采样和深入分析。[8] 使用这种方法,我们已经询问了小鼠CRC模型中的免疫状况,并揭示了它在暴露于不同的TNIKi化合物后如何变化。结合体内和体外的其他研究,结果表明,TNIKi治疗可以通过诱导免疫原性肿瘤细胞死亡来触发强大的CD8 + T细胞介导的抗肿瘤反应,[9]进一步促进CD8 + T细胞
公正的体外和体内药物锚筛网鉴定了MTAP-DEL中MTA合件PRMT5抑制剂的抗性和敏化机制
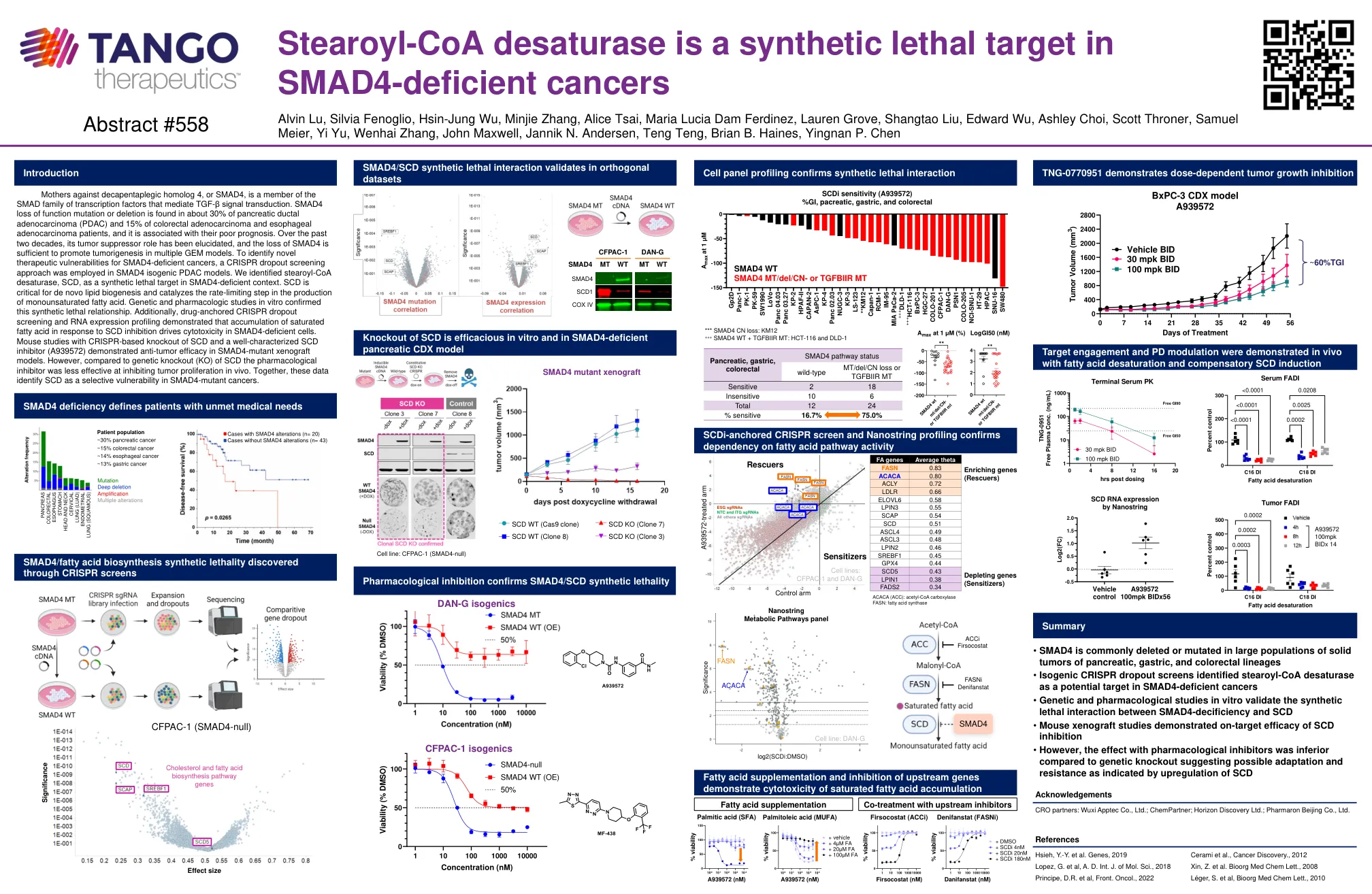
母亲反对脱皮性同源物4(SMAD4)是介导TGF-β信号转导的Smad转录因子家族的成员。SMAD4功能突变或缺失的丧失在约30%的胰腺导管腺癌(PDAC)和大肠癌腺癌和食管腺癌患者的15%,并且与预后不良有关。在过去的二十年中,其肿瘤抑制作用的作用已被阐明,SMAD4的损失足以促进多种GEM模型中的肿瘤发生。为了识别SMAD4缺陷癌的新型治疗脆弱性,在SMAD4等源性PDAC模型中采用了CRISPR辍学方法。我们将stearoyl-COA去饱和酶SCD鉴定为Smad4缺陷型环境中的合成致命靶标。scd对于从头脂质生物发生至关重要,并催化单不饱和脂肪酸的产生速率限制步骤。体外遗传学和药理学研究证实了这种合成的致命关系。此外,药物锚定的CRISPR辍学筛选和RNA表达分析表明,饱和脂肪酸对SCD抑制作用的积累驱动SMAD4缺陷细胞中的细胞毒性。用基于CRISPR的SCD敲除和特征良好的SCD抑制剂(A939572)的小鼠研究表明,SMAD4-突变异种移植模型中具有抗肿瘤功效。但是,与SCD的遗传基因敲除(KO)相比,药理学抑制剂在抑制体内肿瘤增殖方面的有效性较小。一起,这些数据将SCD识别为SMAD4突变癌中的选择性漏洞。

敏捷任务设计实用指南和航天器
抽象的传统上,复杂的空间硬件和任务设计一直是文档驱动的过程[1]。但是,鉴于跨学科设计在动态的全球太空经济中的复杂性日益增长,硬件设计社区正在寻找方法来优化工作流程,鉴于当前工具和流程的挑战和局限性。“简介”通过确定任务生命周期与太空任务分析和设计之间的重叠来提供一些有关太空任务和硬件设计的背景。以下节“敏捷航空航天”提供了有关从传统的顺序模型转移到空间设计和开发中并发和迭代敏捷模型的好处。讨论了成功的敏捷公司和远程工作的兴起的一个例子。“敏捷太空行业中远程团队的实用指南”部分提供了实用的准则,以从敏捷方法的优势中受益,尤其是在远程开发中数据驱动的系统工程方法之后。介绍空间运营的复杂性日益复杂,太空领域的公司和机构正在寻找工作流和开发优化的工具和方法。这与软件行业中发生的情况相当[2,3],在过去的几十年中,必须解决类似的问题。在软件行业的敏捷方式时代,将这些方法带入硬件设计并呈现出数据驱动的系统工程(DDSE)方法的敏捷空间硬件和任务设计的想法。本节介绍了任务生命周期和太空任务分析与设计(SMAD)过程,这些过程与实现空间操作的骨架相互联系。

癌症干细胞和与肿瘤相关的巨噬细胞作为肿瘤进展的伴侣:串扰和晚期生物信息学工具的机制,以剖析其表型和相互作用
癌症干细胞(CSC)是肿瘤质量中的一个小子集,这显着促进了癌症的进展,通过各种致癌途径的失调,促进肿瘤生长,化学抗性和转移形成。CSC的侵略性行为由几种细胞内信号通路,例如Wnt,NF-KAPPA-B,Notch,HydgeHog,Jak-Stat,Pi3K/Akt1/MTOR,TGF/TGF/TGF/SMAD,PPAR,PPAR,PPAR和MAPK激酶,以及诸如外胞外小叶等信号,以及诸如外胞外叶子,以及分类的cy虫,以及分类的分解。趋化因子,促血管生成和生长因子,最终调节CSC表型。在这种情况下,肿瘤微环境(TME)是建立允许性肿瘤生态位的关键参与者,其中CSC与各种免疫细胞进行复杂的通信。“致癌”免疫细胞主要由B和T淋巴细胞,NK细胞和树突状细胞表示。在免疫细胞中,巨噬细胞由于其不同的亚群而表现出更塑性和适应性的表型,其特征在于免疫抑制和炎症表型。Speci fi cally, tumor-associated macrophages (TAMs) create an immunosuppressive milieu through the production of a plethora of paracrine factors (IL-6, IL-12, TNF-alpha, TGF-beta, CCL1, CCL18) promoting the acquisition by CSCs of a stem-like, invasive and metastatic phenotype.tams已经证明了通过直接配体/受体(例如CD90/CD11b,Lsectin/btn3a3,epha4/ephrin)相互作用与CSC进行通信的能力。另一方面,CSC表现出其影响免疫细胞的能力,创造了有利的微环境,以实现癌症的进展。如今,有趣的是,CSC和TME的双向影响会导致表观遗传重编程,从而维持恶性转化。

TGF-β 诱导的 TMEPAI 减弱了三重... 的反应
目的:三阴性乳腺癌 (TNBC) 是一种难治性乳腺癌,预后不良,治疗选择有限。先前的研究表明,TNBC 具有高跨膜前列腺雄激素诱导蛋白 (TMEPAI) 表达。已知 TMEPAI 由 TGF- β /Smad 信号诱导,并具有致瘤功能,可将 TGF- β 从肿瘤抑制因子转化为肿瘤促进因子并诱导上皮 - 间质转化 (EMT)。因此,我们旨在确定 TMEPAI 在 TGF- β 存在下使用几种抗癌药物治疗三阴性乳腺癌细胞中的作用。方法:在三阴性乳腺癌细胞 BT549 中进行 TMEPAI 敲除 (KO)。 TMEPAI 编辑是使用 CRISPR-Cas9 系统开发的,它使用两种 sgRNA 组合来完全去除 TMEPAI 基因的外显子 4。进行基因分型和蛋白质组学分析以检查 TMEPAI-KO 细胞的建立。在 TGF-β 处理的情况下,野生型 (WT) 和 KO 细胞用于确定几种抗癌药物的 50% 抑制浓度 (IC 50 ):阿霉素、顺铂、紫杉醇和比卡鲁胺。结果:通过完全去除 TMEPAI 基因成功建立了 KO 细胞,这在基因组和蛋白质组学分析中得到了证实。此外,在 TMEPAI-KO 细胞中,我们发现阿霉素和紫杉醇的 IC 50 显著降低,而顺铂和比卡鲁胺的影响最小。我们的研究结果表明,TGF-β 诱导的 TMEPAI 减弱了 TNBC 对阿霉素和紫杉醇的反应,但对顺铂和比卡鲁胺的反应没有影响。结论:TGF-β 诱导的 TMEPAI 导致 TNBC 治疗对阿霉素和紫杉醇的反应降低,但对顺铂和比卡鲁胺的影响最小。需要进一步研究以在其他生长因子诱导的细胞以及体内模型中证实我们的发现。关键词:TMEPAI、TGF-β、阿霉素、紫杉醇