XiaoMi-AI文件搜索系统
World File Search SystemSMAD4

不同的内皮机制导致 Alk1 和 SMAD4 功能丧失导致动静脉畸形
。CC-BY 4.0 国际许可(未经同行评审认证)是作者/资助者,他已授予 bioRxiv 永久展示预印本的许可。它是此预印本的版权持有者此版本于 2025 年 1 月 3 日发布。;https://doi.org/10.1101/2025.01.03.631070 doi:bioRxiv 预印本

使用协作交叉小鼠群研究宿主遗传背景对杂合 Smad4 基因敲除体重变化的影响
摘要:肥胖及其伴随疾病已成为全球主要的健康问题,目前肥胖是全球第五大死亡原因。复杂的环境和遗传因素是造成当前肥胖流行的原因。饮食、生活方式、化学物质暴露和其他混杂因素在人类中难以控制。小鼠模型有助于研究遗传性体重增加,因为小鼠的遗传和环境风险因素是可以控制的。对具有各种遗传背景和已建立遗传结构的小鼠品系进行研究,为发现和分析与性状相关的基因组位点提供了无与伦比的机会。在本研究中,我们使用了协作杂交 (CC),一种大型重组近交系小鼠品系,使用 CC 小鼠的杂合 Smad 4 敲除谱进行预测研究,以了解和有效识别体重增加的倾向。雄性 C57Bl/6J Smad4+/− 小鼠与来自 10 个不同 CC 品系的雌性小鼠交配,产生 F1 小鼠 (Smad4+/− x CC)。每周测量一次体重 (BW),直至第 16 周,然后每月测量一次,直至研究结束(第 48 周)。评估并呈现所评估特征的遗传力 (H2)。对用于预测小鼠体重变化和基因型的各种机器学习算法进行了比较分析。我们的数据显示,在实验过程中,不同 CC 品系的 F1 小鼠的体重记录在野生型和突变型 Smad4 小鼠之间有所不同。遗传背景会影响体重增加,在杂合 Smad4 敲除的情况下,一些品系体重增加更多,而其他品系体重增加较少,但总的来说,除了少数品系外,突变会导致小鼠超重。在对照组和突变组中,雌性 %BW 的遗传力 (H2) 值高于雄性。此外,具有野生型基因型的两种性别都比突变组表现出更高的遗传力值。逻辑回归使用机器学习提供最准确的小鼠基因型预测。我们计划在更多 CC 品系和每品系小鼠上验证所提出的方法,以扩大机器学习用于 BW 预测的文献。
公正的体外和体内药物锚筛网鉴定了MTAP-DEL中MTA合件PRMT5抑制剂的抗性和敏化机制
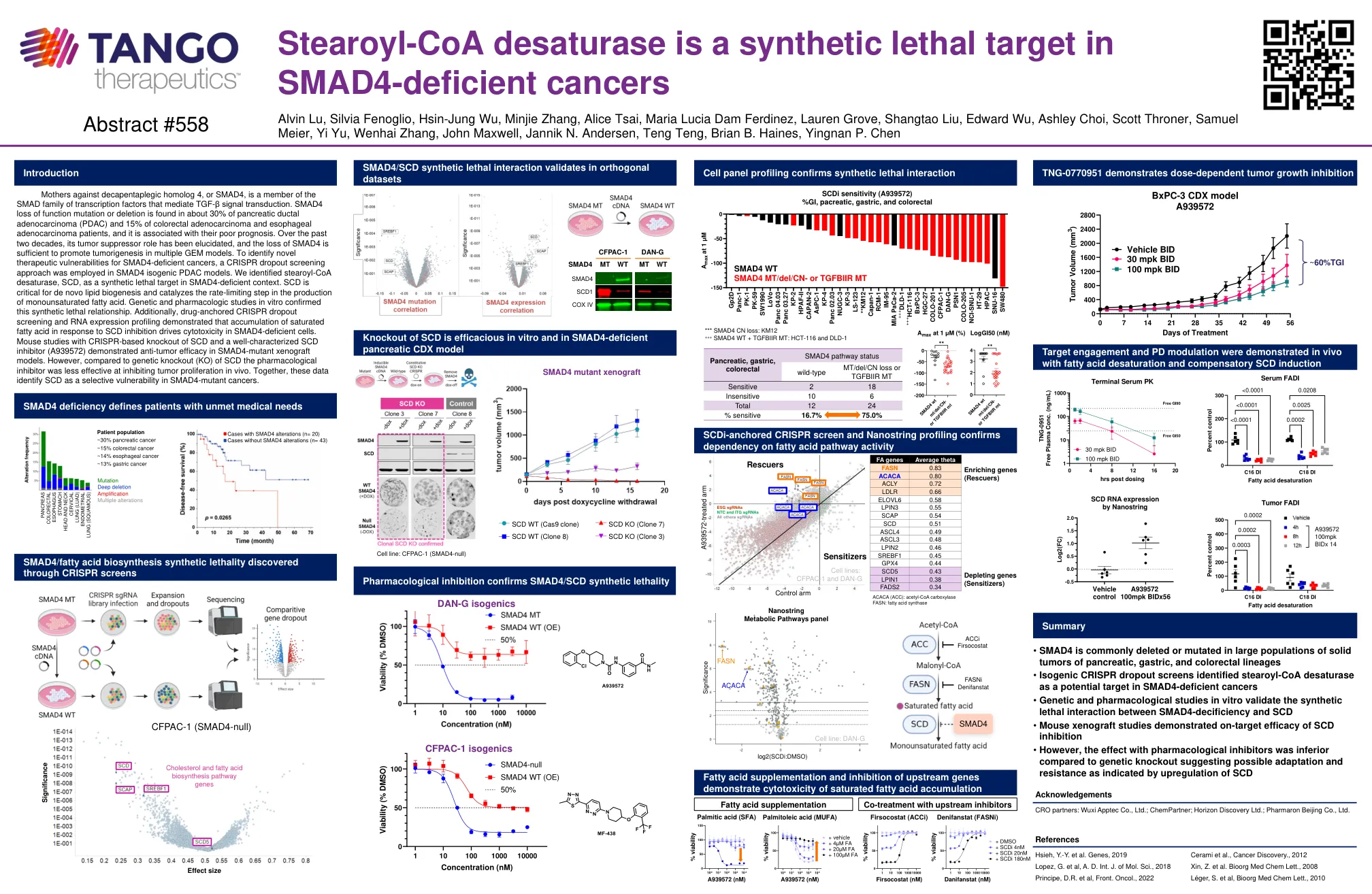
母亲反对脱皮性同源物4(SMAD4)是介导TGF-β信号转导的Smad转录因子家族的成员。SMAD4功能突变或缺失的丧失在约30%的胰腺导管腺癌(PDAC)和大肠癌腺癌和食管腺癌患者的15%,并且与预后不良有关。在过去的二十年中,其肿瘤抑制作用的作用已被阐明,SMAD4的损失足以促进多种GEM模型中的肿瘤发生。为了识别SMAD4缺陷癌的新型治疗脆弱性,在SMAD4等源性PDAC模型中采用了CRISPR辍学方法。我们将stearoyl-COA去饱和酶SCD鉴定为Smad4缺陷型环境中的合成致命靶标。scd对于从头脂质生物发生至关重要,并催化单不饱和脂肪酸的产生速率限制步骤。体外遗传学和药理学研究证实了这种合成的致命关系。此外,药物锚定的CRISPR辍学筛选和RNA表达分析表明,饱和脂肪酸对SCD抑制作用的积累驱动SMAD4缺陷细胞中的细胞毒性。用基于CRISPR的SCD敲除和特征良好的SCD抑制剂(A939572)的小鼠研究表明,SMAD4-突变异种移植模型中具有抗肿瘤功效。但是,与SCD的遗传基因敲除(KO)相比,药理学抑制剂在抑制体内肿瘤增殖方面的有效性较小。一起,这些数据将SCD识别为SMAD4突变癌中的选择性漏洞。

stearoyl-COA去饱和酶是SMAD4缺陷癌症中的合成致命靶标
与功能基因组学研究结合的大规模DNA测序在表征癌症基因组方面起着关键作用,揭示了缺失事件的重要性,这些事件的重要性通过肿瘤抑制基因的丧失来促进肿瘤生长。诸如癌症基因组图集计划(TCGA)之类的倡议提供了整个人类癌症遗传改变的综合图,表明缺失事件通常延伸到肿瘤抑制基因基因座,从而导致相邻基因的代码。尽管这些乘客事件可能不会赋予肿瘤的直接健身优势,但它们可以创建可以通过治疗剥削的副脆弱性。一个例子是由甲基腺苷磷酸化酶(MTAP)丧失赋予PRMT5抑制作用的附带脆弱性,该基因经常与描述良好的肿瘤抑制基因CDKN2A相关。1-3 MTAP编码蛋白质MTAP,蛋白MTAP是蛋氨酸拯救途径中的临界酶,该过程从多胺合成的副产物中循环蛋氨酸,甲基噻吩腺苷(MTA)。CDKN2A的丧失发生在所有人类癌症中的10-15%中,并且在组织学上的频率更高,例如恶性周围神经鞘肿瘤,胶质母细胞瘤(GBM),间皮瘤,间皮瘤,尿路上皮癌,食管鳞状细胞癌,胰腺癌,胰腺腺瘤腺瘤,<- <- <-

图S8:RAB10定位的确定
要检查RAB10的定位是否在SMAD4损失的设置中发生变化,我们在SW620和HT29的SMAD4中存在或不存在的HT29同源细胞上进行了免疫荧光实验。将带有诱导质粒PSMAD4的SMAD4阴性细胞系HT29和SW620在第0天接种,在第1天用强力霉素处理72H。rab10(#ab237703 1:400)随后与早期(EEA1,#BD 610456 1:400)和迟到(CD63,#AB1318 1:400)共同染色。用Zeiss Axio观察者荧光显微镜拍摄的图片。

多路复用时分辨荧光共振能量转移超高通量筛选测定法,用于靶向SMAD4 – SMAD3 – DNA复合物
转化的生长因子-BETA(TGFβ)信号通路在建立免疫抑制性肿瘤微环境中起着至关重要的作用,使抗TGFβ剂成为癌症免疫疗法的重要领域。然而,针对上游细胞因子和受体的当前抗TGFβ药物的临床翻译仍然具有挑战性。因此,小分子抑制剂的发展特异性靶向TGFβ途径的下游主调节器SMAD4,将采取一种替代方法,具有明显的抗TGFβ信号传导的替代方法。在这项研究中,我们介绍了在超高通量筛选(UHTS)1536孔板格式中基于细胞裂解物的多路复用时间分辨荧光共振能量转移(TR-FRET)测定。该测定法可以同时监测SMAD4和SMAD3之间的蛋白质 - 蛋白质相互作用,以及SMADS及其共识DNA结合基序之间的蛋白质-DNA相互作用。多路复用的TR-FRET分析表现出高灵敏度,从而使单氨基酸分辨率下的Smad4-Smad3-DNA复合物进行了动态分析。此外,多路复用的UHTS分析证明了筛选小分子抑制剂的鲁棒性。通过对FDA批准的生物活性化合物库进行试验筛选,我们将gambogic Acid和Gambogenic Acodic鉴定为潜在的HIT化合物。这些概念验证的发现强调了我们优化的多重TR-FRET平台的大规模筛选的实用性,以发现针对SMAD4-SMAD3 – DNA复合物作为新型抗TGFβ信号剂的小分子抑制剂。



由...
结直肠癌(CRC)是全球最致命,通常被诊断出的肿瘤之一。几个基因参与其发展和进展。最常见的突变涉及APC,KRAS,SMAD4和TP53基因,这表明CRC依赖于相关途径的伴随改变。但是,使用经典的分子方法,同时分析这些途径之间的互连并不容易。为了克服这一局限性,最近这些途径已包括在一个巨大的化学反应网络(CRN)中,描述了健康的结直肠细胞处理如何通过生长因子从环境中感知的信息。从此CRN开始,我们提出了一个计算模型,该模型模拟了全局信号网络对单个或多个并发突变引起的效果。该模型已在三种情况下进行了测试。首先,我们通过APC,KRAS,SMAD4或TP53中的突变量化了网络蛋白浓度所引起的变化。第二,我们计算了由多达两个影响网络上游蛋白的并发突变引起的p53浓度的变化。第三,我们考虑了受KRAS功能增长影响的突变细胞,并模拟了Dabrafenib的作用,表明该模型可用于确定将最有效的药物传递到细胞中。通常,所提出的方法显示出几个优点,因为它允许量化由单个或多个给定突变引起的蛋白质浓度的变化。此外,可以使用CRC全局信号网络的模拟来识别新的治疗靶标,或者披露所涉及途径之间的意外相互作用。

E3泛素连接酶ASB8促进selinexor诱导的蛋白酶体降解XPO1
Selinexor (KPT-330) 是一种具有强效抗癌活性的 Exportin-1 (XPO1, CRM1) 小分子抑制剂,最近已获得 FDA 批准用于治疗复发/难治性多发性骨髓瘤和弥漫性大 B 细胞淋巴瘤 (DLBCL),目前正在对许多其他适应症进行临床研究。由于 selinexor 与其他药物(尤其是硼替佐米和地塞米松)联合使用时经常表现出协同作用,因此采用更全面的方法来发现新的有益相互作用将具有重要价值。此外,对患者进行分层、个性化治疗和改善临床结果需要更好地了解药物反应背后的遗传脆弱性和耐药机制。在这里,我们使用 CRISPR-Cas9 功能丧失化学遗传学筛选来识别慢性粒细胞白血病、多发性骨髓瘤和 DLBCL 细胞系中 selinexor 与药物基因的相互作用。我们发现 TGF β -SMAD4 通路是多发性骨髓瘤细胞对 selinexor 耐药的重要介质。此外,该通路活性较高与接受 selinexor 治疗的多发性骨髓瘤患者的无进展生存期延长相关,这表明 TGF β -SMAD4 通路是预测治疗结果的潜在生物标志物。此外,我们还发现 ASB8(锚蛋白重复序列和 SOCS 盒含 8)是所有测试癌症类型中 selinexor 敏感性的共同调节剂,ASB8 敲除和过表达都会导致 selinexor 过敏。从机制上讲,我们表明 ASB8 促进了 selinexor 诱导的蛋白酶体降解 XPO1。这项研究深入了解了影响 selinexor 治疗反应的遗传因素,并可以支持预测性生物标志物和新药物组合的开发。