XiaoMi-AI文件搜索系统
World File Search SystemSNPs

ACE基因多态性和约旦成年人口中高血压的易感性
高血压是与遗传和环境风险因素相关的最常见和最复杂的疾病之一。血管紧张素转换酶(ACE)在肾素 - 血管紧张素系统途径中很重要。ACE的基因表达已被研究为可能的高血压标记。这项研究调查了约旦人口中ACE1中的多态性与ACE2基因的多态性与高血压敏感性之间的关联。该研究总共包括200名高血压患者和180个健康控制。进行了聚合酶链反应(PCR),以基因型的基因型ACE1GENE的候选多态性(RS4646994)。使用ACE1和ACE2基因的Luminex DNA阵列技术用于基因分型SNP(RS4359,RS4344,RS4341,RS4343和RS2106809)。我们的发现表明,SNP与高血压之间没有有关理和基因型频率的关联。然而,rs4359与饮食(PP = 0.049),知识HTN(P = 0.042)和DM年数(P = 0.003)显着相关。rs4341与饮食(p = 0.032),周围血管疾病(p = 0.005)和慢性肾脏病有关(p = 0.049)。虽然rs4343与饮食(p = 0.031),糖尿病(p = 0.032)和其他药物(p = 0.025)相关。此外,ACE1基因的四个SNP的单倍型与HTN患者和健康对照没有显着关联。我们的发现表明,ACE基因中的多态性与约旦成年人口的过度张力发展的风险之间没有关联。

英国生物样本库对早期全基因组关联研究中发现的全基因组显著信号的可重复性
摘要 随着大型生物库的建立,与各种表型相关的单核苷酸多态性 (SNP) 的发现速度加快了。一个悬而未决的问题是,早期全基因组关联研究 (GWAS) 中确定的具有全基因组意义的 SNP 是否也会在后来在生物库进行的 GWAS 中得到复制。为了解决这个问题,作者检查了一个公开的 GWAS 数据库,并确定了同一表型的两个独立 GWAS(早期的“发现” GWAS 和后来在英国生物库进行的复制 GWAS)。该分析评估了来自 4,397,962 名参与者的 9 种表型的 136,318,924 个 SNP(其中 6,289 个在发现 GWAS 中达到 p<5e-8)。总体复制率为 85.0%,二元表型的复制率低于定量表型(分别为 58.1% 和 94.8%)。二元表型的 SNP 效应大小下降了 18.0%,但定量表型的 SNP 效应大小增加了 12.0%。利用发现的 SNP 效应大小、表型特征(二元或定量)和发现的 p 值,我们建立并验证了一个模型,该模型预测 SNP 复制,受试者工作曲线下面积 = 0.90。虽然无法复制通常可能反映出缺乏效力,而不是真正的假阳性结果,但这些结果提供了关于哪些发现的关联可能在后续的 GWAS 中再次出现的信息。

学术界的气候意识:奥地利国际学生的空中旅行态度的研究
现代技术使使用基因组数据可以预测和自定义预防和治疗疾病的策略。人类基因组中存在数百万个单核苷酸多态性(SNP),全基因组关联研究(GWAS)有助于识别SNP与各种疾病之间的关联联系(1)。经常具有较弱的个体影响的多态性可能会集体与疾病表现出很强的相关性(2)。多基因风险评分(PRS)是一种线性回归模型,该模型使用了带有GWAS的权重的单个SNP,传统上已被用来评估多因素疾病表现的风险。尽管PRS由于其简单性和良好的预测能力而正确地成为了最受欢迎的工具,但它具有重大局限性,例如无法说明上静脉的非线性效应。尽管从历史上看,该术语已用于描述各种遗传事件,但最合适的定义是Fisher(3)提出的。这是统计上的上述,它是指遗传变异对疾病的影响的现象。epitsisis是一个积极研究的领域,已经被证明对多种疾病产生了重大影响(4)。上位性是建立可靠的多基因风险模型的一个挑战性方面,因为线性方法通常不足以捕获遗传变异和疾病之间的非线性关系。

根据...
2型糖尿病(T2D)的发病机理可能随着年龄的增长而改变。在这里,我们使用了基于诊断年龄的层次化,以深入了解不同年龄段的T2D遗传学和因果风险因素。我们根据诊断年龄(<50、50-50-60、60-60-60-70和70岁)(总计24,986例),对T2D和T2D亚组进行了全基因组关联研究(GWAS)。作为对照对象,参与者在随访结束时至少70岁,而没有发展T2D(n 5 187,130)。GWAS识别208独立的铅single核苷酸多态性(SNP)映射到与T2D相关的69个基因座(p <1.0e 8)。除其他外,映射到CDKN2B-AS1的SNP和映射到TCF7L2的多个独立的SNP与70岁后诊断的病例相比,比在50岁之前诊断出的病例更为密切。基于不同的病例组,我们进行了两样本的孟德尔式统治。最值得注意的是,我们观察到了被认为的风险因素,BMI和T2D之间的关联随诊断年龄的增加而减弱。总体而言,我们的结果表明,基于诊断年龄的T2D的地层揭示了亚组特异性遗传学和因果决定因素,支持

整合全基因组关联研究和表达数量性状基因座定位的正向遗传学方法来解析玉米(Zea mays)叶片的发育
玉米 (Zea mays) 叶片发育的遗传基础表征可支持育种工作,以获得具有更高活力和生产力的植物。在本研究中,对 197 个双亲和多亲本玉米重组自交系 (RIL) 的映射面板在苗期对多种叶片性状进行了分析。使用 RNA 测序来估计 RIL 中 29 573 个基因模型的转录水平并得出 373 769 个单核苷酸多态性 (SNP),然后结合这些数据使用正向遗传学方法来精确定位参与叶片发育的候选基因。首先,将叶片性状与基因表达水平相关联以确定转录本 - 性状相关性。然后,在全基因组关联 (GWA) 研究中将叶片性状与 SNP 相关联。采用表达数量性状基因座映射方法将 SNP 与基因表达水平相关联,并根据转录本 - 性状相关性和 GWA 确定候选基因的优先顺序。最后,进行了网络分析,将所有转录本聚类到 38 个共表达模块中。通过整合正向遗传学方法,我们确定了 25 个高度富集特定功能类别的候选基因,为液泡质子泵、细胞壁效应器和囊泡交通控制器在叶片生长中的作用提供了证据。这些结果解决了叶片性状确定的复杂性,并可能支持玉米的精准育种。

一般基因检测,躯体疾病 AHS - M2146
背景 如果基因突变不在生殖系内(即配子内),则称为“体细胞突变”;因此,这些突变不会从父母传递给后代。体细胞突变可能从头出现或生命后期出现,在肿瘤中非常常见(Raby & Blank,2022 年)。体细胞突变有很多种类型,包括单核苷酸多态性 (SNP);结构变异,如缺失、倒位或易位,以及较小的染色体异常,如短串联重复或基因融合。大多数突变不会导致疾病(Kohlmann & Slavotinek,2022 年)。SNP 是最常见的基因突变类型,包括错义突变。这些突变是单个碱基对的变化,其中一个核苷酸取代了另一个核苷酸。超过 65% 的由基因突变引起的疾病是由于 SNP 引起的(Kohlmann & Slavotinek,2022 年)。根据全基因组测序的估计,任何给定个体的平均 SNP 数量为 280 万到 390 万 (Kohlmann & Slavotinek, 2022)。插入/缺失 (Garrett 等人) 多态性通常为单个核苷酸,但可能多达四个核苷酸。SNP 通常会导致移码突变,从而导致过早终止密码子和等位基因失效 (Kohlmann & Slavotinek, 2022)。结构变异通常被归类为大于 1000 个碱基对。这些包括缺失、重复、倒位、易位或环状染色体形成。由于受影响的基因数量众多,这些变异通常会导致严重的遗传异常;例如,慢性粒细胞白血病的主要原因是由于 9 号和 22 号染色体之间的易位,导致融合基因。最常见的结构变异是拷贝数变异 (CNV),指的是不同个体中 DNA 片段拷贝数的不同。例如,一个人可能有三个特定片段的拷贝,而另一个人可能只有两个。这些变异可能导致受影响基因的失调、功能获得或功能丧失 (Kohlmann & Slavotinek, 2022)。需要或产生精确数量蛋白质产品的敏感基因往往更容易受到这些变异的影响 (Bacino, 2022)。任何大小的突变都可能是致病的,必须根据突变导致疾病的可能性进行分类。美国医学遗传学和基因组学学院 (ACMG) 将突变分为五类,如下:致病、可能致病、意义不明、可能良性和良性。 “可能致病”和“可能良性”指的是比其各自的致病和良性类别更弱的证据,“意义不确定”指的是证据不符合良性或致病性的标准,或双方的证据相互矛盾(Kohlmann & Slavotinek,2022)。预测算法已用于解释变异并预测变异是否会影响基因功能或基因剪接。这些算法是公开的
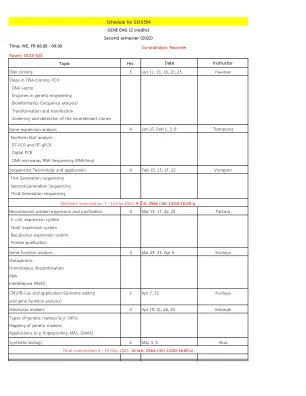
2310554 Gene Eng的时间表(2个学分)第二...
分子标记物4月4日,21,26,28 Veerasak类型的遗传标记(例如SNP)遗传标记应用的映射(例如指纹,MAS,GWAS)合成生物学,5月3日,5 Alisa最终考试8-19 2023年5月19日:16พ.2566เวลา13:00-16:00น。

国际寄生虫学杂志
Haemonchus contortus是小型反刍动物中最致病的线虫,而驱虫抗性(AR)阻碍了其有效的控制。需要早期检测AR状态才能减少AR的选择,并且无法使用表型测试来实现。对于苯二唑唑(BZ),在同种型1β-微型蛋白基因中以单核苷酸多态性(SNP)为特征的AR相关等位基因的检测允许Stron Gyles的早期AR检测。在抗BZ的种群中已经描述了F200Y,F167Y,E198A和E198L多态性,区域之间的频率有明显变化。一种新型的数字PCR(DPCR)可以检测H. contortus中所有上述多态性。测定进行了验证。然后,分析了26个奥地利人和10个意大利绵羊农场的幼虫,并在农场一级合并。对于所有测定,证明了15份/μL电阻等位基因的检测极限和高度准确性,从而可以在大多数样品中检测1%的等位基因频率。在奥地利的样本中,在所有农场都检测到了F200y等位基因的频率升高。第一次在奥地利的H. tortus中发现了密码子167和密码子198中的多态性。在意大利样品中,电阻等位基因的频率仍然相对较低,但F200Y抗性等位基因可追溯。总而言之,我们首次开发了DPCR分析,该测定目标是针对H. contortus中与BZ抗性相关的所有相关性SNP。对AR开发的未来研究可能会受益于基于SNP的监视,其中包括所有相关性SNP的开发测定法。改进的监视将包括其他重要的,尽管病原体较少的线虫属。

勘误表:高效基因编辑策略......
图 1 | 葡聚糖水二激酶 (GWD) 1 — gRNA 靶区的结构和完整等位基因序列。上图为外显子(方框)和包含碳水化合物结合模块 (CBM) 的区域的整体基因结构。左图:外显子 1 和内含子的核苷酸序列。右图:外显子 24 和 25,包括内含子。外显子以大写字母表示,并标明氨基酸序列。SPUD 数据库中包含的品种的小核苷酸多态性 (SNP) 以红色标记,Saturna 中发现的 SNP 以下划线表示。灰色箭头表示 gRNA(gA、gB、gC、gD、gE、gI、gJ、gK、gL 和 gM),其中 PAM 位点以粗体标记。红色箭头表示诊断性 IDAA PCR 引物。 “ CFATC ” 区域含有半胱氨酸,据推测该区域参与二硫键间或二硫键内形成,因此推测参与 GWD 活性的氧化还原状态调节,该区域以粗体标记。活性位点组氨酸残基也以粗体标记。

BRC生物信息学Illumina基因分型QC和Imputaion Service Pipeline
来自Illumine基因分型微阵列的原始基因分型数据以图像格式(IDAT文件)上传到Genomestudio软件中。此软件通过基于荧光强度的相似性来确定样品的自动聚类来确定等位基因的身份。但是,由于异常强度模式,默认聚类算法可能无法识别有效的簇,也可以将错误的基因型分配给样品。可以通过手动审查和回忆SNP来解决数据的可靠性,信心和整体质量,从而使其成为非常关键的质量控制(QC)程序,然后再使用PLINK或遗传解释进一步QC。此过程可以按; - =几周,具体取决于项目的大小(SNP的数量)。GenomeStudio软件需要至少提供的VWW样品来准确地群集数据,并且如果样本超过= www,我们必须在批处理中进行,以便花费更多的时间。此外,更大的芯片也需要更多的时间,因此成本更高。