XiaoMi-AI文件搜索系统
World File Search SystemSNV

博士课程课程单元英语面额...
课程单元目录1。序列分析 - 了解DNA序列,序列相似性,身份和同源性的基本概念,数据库搜索:BLAST,FASTA,FASTA和其他序列分析工具分配同源性。底漆设计,PCR和Sanger序列分析。2。转录组分析 - RNA-seq数据分析中的概念:数据预处理和数据处理步骤:映射算法,例如BWA和BOWTIE2;使用RNA-seq数据,统计方法,各种平台的相对优点进行差异基因表达分析。下游验证的底漆设计。从RNA-seq数据中测量基因,lncRNA,siRNA。3。微生物组分析-16S rRNA数据分析,基于比对的聚类/系统发育树,基于组成的聚类。基于数据库,主组件分析和其他聚类工具的注释。4。SNP分析 - 靶基因或整个基因组,基因预测算法,变体的鉴定 - SNP/SNV的鉴定。基因组广泛关联研究背后的概念。介绍各种
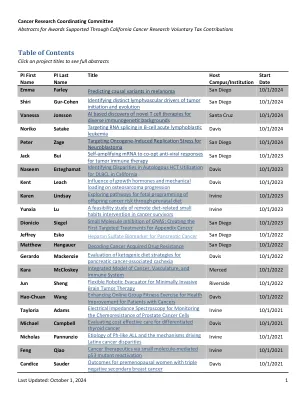
目录
预测黑色素瘤的致病变异 校园:UCSD 首席研究员:Emma Farley 开始日期:2024 年 10 月 1 日 结束日期:2025 年 9 月 30 日 金额:95,000 美元 摘要:癌症的各个方面都由基因表达的变化驱动。增强子是控制特定基因或基因表达的时间、位置和水平的基因组元素,因此,增强子提供基因表达的指令。虽然人们广泛关注蛋白质编码变异和改变蛋白质编码区域的基因组变化,但增强子变异如何促进癌症的发生、进展、转移和对治疗的反应却研究甚少。增强子包含与包括癌症在内的疾病相关的大多数变异,但精确定位致病变异是一项重大挑战,因为它们通常嵌入在大量惰性变异中。我们知识上的这一空白阻碍了充分利用基因组数据的潜力来了解和治疗癌症的努力。清楚了解哪些增强子变异对癌症的各个方面有影响对于了解癌症发生和发展的遗传基础、开发新型疗法、改善诊断和对患者进行分层以进行更有针对性的治疗至关重要。在这项研究中,我们将试行一种新方法来识别导致黑色素瘤基因表达变化的增强子变异。我们是识别增强子中因果变异的专家,这些变异会在发育中的胚胎中改变基因表达和细胞身份。我们发现,低亲和力结合位点对于精确控制基因表达至关重要。增强子中低亲和力位点的普遍使用造成了基因组中的脆弱性,SNV 可以增加增强子中结合位点的亲和力,从而导致功能基因表达获得,从而改变细胞身份。我们已经在心脏和肢体发育的背景下证明了这一点。我们现在希望应用这些知识来深入了解导致癌症各个方面的因果变异。我们计划最初重点关注黑色素瘤的转移、免疫疗法反应和耐药性。在我们的初步研究中,我们已表明,在体细胞黑色素瘤 eQTL 中发现的亲和力优化 SNV 会增加 DAAM1 的表达并增加细胞迁移。成功完成该项目将揭示增强子变体对癌症进展和治疗的贡献。

动物和怀疑的农作物串,环境,…
课程单元目录1。序列分析 - 了解DNA序列,序列相似性,身份和同源性的基本概念,数据库搜索:BLAST,FASTA,FASTA和其他序列分析工具分配同源性。底漆设计,PCR和Sanger序列分析。2。转录组分析 - RNA-seq数据分析中的概念:数据预处理和数据处理步骤:映射算法,例如BWA和BOWTIE2;使用RNA-seq数据,统计方法,各种平台的相对优点进行差异基因表达分析。下游验证的底漆设计。从RNA-seq数据中测量基因,lncRNA,siRNA。3。微生物组分析-16S rRNA数据分析,基于比对的聚类/系统发育树,基于组成的聚类。基于数据库,主组件分析和其他聚类工具的注释。4。SNP分析 - 靶基因或整个基因组,基因预测算法,变体的鉴定 - SNP/SNV的鉴定。基因组广泛关联研究背后的概念。介绍各种

技术评估提交清单和问卷 (GEN-CQD-003-v4)
热点;这些检测旨在描述肿瘤的基因组组成,并有助于识别疾病的潜在机制以指导临床决策。这些测试不仅包括单个相关基因的突变,还包括已建立的癌症途径中相关基因的突变模式,并且通常包括对整体突变负担的评估。这些测试通常涉及对感兴趣基因的整个外显子区域的测序(在综合基因组或全外显子组测序中),并且还可能包括选定的内含子区域。CGP 可以在一次检测中检测多种类型的分子改变(即 SNV、小和大 INDEL、拷贝数改变 (CNA)、结构变异 (SV) 和剪接位点变异)。跨多个基因观察到的突变模式可用于推断临床相关病因,例如 DNA 错配修复缺陷和微卫星不稳定性,并且可以确定总突变负荷/负担 (TMB)。 CGP 测试还可能包括 RNA 测序以检测结构变异,例如易位或大量缺失,并检测功能性剪接突变。

Physiol. Res. 72 (Suppl. 3): S267-S275, 2023 https://doi.org/10.33549/physiolres.935154 综合基因组分析在 C 预测测试中的应用
斯洛伐克共和国。电子邮件:svoboda22@uniba.sk 简介 个性化医疗 (PM) 是一种医疗护理模式,它允许根据疾病的分子基础定制医疗保健、治疗和诊断程序。该模型通常使用实验室测试,重点是基于全面的分子和遗传分析选择适当且最佳的治疗方法。个性化医疗在肿瘤学领域最重要的方面之一是识别合适的分子生物标志物,可用于评估预后和预测创新生物治疗的靶点 [1-3]。有关个体肿瘤分子特征的信息有助于肿瘤学家补充传统方法,例如组织定位和肿瘤组织学。在当前的临床实践中,个性化医疗主要以基于单基因的检测形式使用。然而,这种方法正逐渐被下一代测序 (NGS) 所取代 [4]。使用 NGS 分析肿瘤 DNA 可以同时评估数十到数千个基因,从而创建全面的基因组图谱 (CGP)。使用综合基因组比分析单个基因有几个优势,因为它能够同时检测已知驱动恶性生长的所有类型的基因组改变,如单核苷酸变异 (SNV)、插入和

标题:肠道菌群脑瘤诱导的肠道菌群失调调节1
体细胞变体检测是癌症基因组学分析的组成部分。尽管大多数方法都集中在短阅读测序上,但长阅读技术现在在重复映射和变体相位方面具有潜在的优势。我们提出了一种深度学习方法,一种深度学习方法,用于从短读和长阅读数据中检测体细胞SNV,插入和缺失(indels),具有用于全基因组和外显子组测序的模式,并且能够以肿瘤正常,唯一的肿瘤正常,ffpe pppe的样本进行运行。为了帮助解决公共可用培训的缺乏和基准测试数据以进行体细胞变体检测,我们生成并公开提供了一个与Illumina,Pacbio Hifi和Oxford Nanopore Technologies的五个匹配的肿瘤正常细胞线对的数据集,以及基准的变体。在样本和技术(短读和长阅读)中,深度态度始终优于现有呼叫者,特别是对于Indels而言。

genepi:用于整个基因组测序分析的GPU增强的下一代生物信息学管道
驱动了对高级计算基础架构进行分析这些大数据集的需求。这项工作的目的是引入一条创新的生物信息学管道,名为Genepi,以进行WGS简短配对读数的有效和精确分析。构建在具有模块化结构的NextFlow框架上,Genepi结合了GPU加速算法并支持多种工作流程配置。管道可自动从生物学WGS数据中提取生物学相关的见解,包括:与疾病相关的变体,例如单核苷酸变体(SNV),小插入或缺失(Indels),拷贝数变体(CNV)和结构变体(SVS)。针对高性能计算(HPC)环境进行了优化,它利用了工作 - 安排的提交,并行处理以及为每个分析步骤量身定制的资源分配。对合成数据集进行了测试,Genepi准确地识别了基因组变量,并且具有与最新工具相当的性能。这些功能使Genepi成为研究和临床环境中大规模分析的宝贵工具,这是朝着建立国家计算和技术医学中心的关键一步。

迷你招架市场报告
我们还感谢那些接受采访并提供了参与这项研究的人:本杰明·卡尼尔(Benjamin Curnier)(Band and Tatia Lemondzhava(世界银行)(世界银行),Dennis Nderitu(Geappe)(Geappe),Jessica Steifler(Miga),Veit Gohringer,Veit Gohringer,Veit Gohringer,Veit Gohringer, Jasper Haerg,Soumana Kaileu和Carlos Miro(Giorgia Pasqualetto,Ben Hartley,Ingrid Rohrer,Anita Otubu,Nishant Narayan,Dolapo Oluis Tavernier和Abdul Yakubu(Seforall) Pigaht和Rebecca Symington(Get.Invest Finance Catalyst),Agustin Carbo和Carolina Ins Pan(所有),Carol Zulu(UNDP),Miriam Atuya和Tombo Banda(Crossboundary Innovation Lab),Frank Bergh(NRECA),Debajit Palit(印度NTPC商学院),Elena Van Hove(ASU),Stewart Hicks(Bamboo Capital),Jean-Denis Collin(Ecole Poindexter(Ecole Poindexter(Enecole Poingicity)(Enecole Poingicity)(Cliffford Aron),Grreenmax Aron(Grreenmax Capital)访问),詹姆斯·托德(James Todd)(Oikocredit),莎拉·亚历山大(SARAH ALEXANDER(SNV),TORSTEN SCHREIBER(非洲Greentech),Kule Hamilton(Nuru Energy)(Nuru Energy),Trey Jarrarard(Renewvia Solar Africa),Idris Tayebi(Winch Energy) Tecnoambiental),Olivier Dumont(1pwafrica),Vijay Dongare(乌干达糖公司有限公司),Omozaphue Akaalumhe(Privida Energy)和Ignatius Anayawa(GEI Power)。

能源转型分类
我们感谢以下外部审阅人员的宝贵贡献:Salifu Addo(加纳能源委员会)、Abeer Al-Aysah(阿联酋联邦竞争力和统计局)、Edi Assoumou(法国巴黎高科矿业学院)、Edito Barcelona(亚太能源研究中心)、Alessandro Bigazzi(英国能源安全和净零排放部)、Molato Celina(莱索托统计局)、Anjali DeAbreu-Kisoensingh(苏里南统计总局)、Fernando Diaz Alonso(欧盟统计局)、Almirante Dima(莫桑比克矿产资源和能源部、计划与合作局)、Zuzana Dobrotková(世界银行)、Manfred Gollner(奥地利统计局)、Stefan Gsänger(世界风能协会)、Ana Kojakovic(联合国粮食及农业组织)、Louis-Marie Malbec(法国 IFPEN)、 Saroj Rai (SNV)、Pablo Ronco (阿根廷能源部长)、Christoph Rouhana (联合国西亚经济社会委员会)、Behrang Shirizadeh (德勤,法国)、Leonardo Souza (联合国统计司)、Florian Steierer (联合国欧洲经济委员会)、Marek Sturc (欧盟统计局)、Adonay Urrutia (Dirección) General de Energía,Hidrocarburos y Minas,萨尔瓦多),Elizabeth Waters(英国能源安全和净零部)。

参考材料8391
(HG-002)此参考材料(RM)用于验证,优化和过程评估目的。它由一个来自个人基因组项目(ID HUAA53E0)的东欧Ashkenazi犹太血统的男性全人基因组样本组成,可以用来评估基因组测序的变体呼叫的性能。RM 8391的单位由一个含有人类基因组DNA的小瓶组成,该小瓶从单一的大生长人类淋巴母细胞系GM24385(标记为HG-002)中提取,从科里尔医学研究所(Camden,NJ)中提取。小瓶含有大约10 µg的基因组DNA,DNA在Te缓冲液中(10 mM Tris,1 mM EDTA,pH 8.0)。该材料旨在通过获得真实阳性,假阳性和假阴性的估计来评估人类基因组测序变体的性能。测序应用可以包括整个基因组测序,整个外显子组测序以及更多靶向测序,例如基因面板。该基因组DNA旨在以与实验室处理和分析提取的DNA相同的方式进行分析。由于RM是提取DNA的,因此它对于评估诸如DNA提取等分析前步骤没有用,但是它确实挑战了测序库制备,测序机以及映射,对齐和变体调用的生物信息学步骤。此RM并非旨在评估随后的生物信息学步骤,例如功能或临床解释。信息值:为单核苷酸变化(SNV),小插入和缺失(Indels)和纯合参考基因型提供信息值。v3.3.2基准集覆盖了GRCH37组件的88%,使用参考文献1中描述的方法。一个信息值被认为是对RM用户感兴趣和使用的值,但是信息不足以评估与该值相关的不确定性。我们使用当前可用的数据和方法来描述和传播对基因型的最佳,最自信的估计。随着新的数据积累,测量和信息学方法的可用,这些数据和基因组特征将随着时间的流逝而保持。HG-002的数据可以在国家生物技术信息中心(NCBI)序列读取存档中的BioSample SAMN03283347下找到。信息值作为一个变体调用文件(VCF),其中包含基准SNV和小indels,以及描述基准区域的TAB划分的“床”文件,其中任何其他变体在基准VCF中没有任何其他变体都应是错误。信息值不能用来建立计量学可追溯性。本报告中引用的文件可在国家生物技术信息中心(NCBI)托管的FTP站点的基因组中获得。基准VCF和基准区域的FTP站点中的基因组是: