XiaoMi-AI文件搜索系统
World File Search Systemnucleotides
DNA和RNA结构,蛋白质相互作用,损伤和...
•主要凹槽的访问和潜在相互作用的访问和数量要比较小的凹槽•对于129个DNA结合蛋白中的大多数,〜2/3的触点是范德尔壁,均为her walls,静止为H键(有或没有中间水)和几个离子P Backbone/arg/arg,lys或n terminus。•蛋白-DNA界面平均涉及24个残基和12个核苷酸•蛋白质结合可以伴随DNA“诱导拟合”的扭转,这也可以为不同的蛋白质或同一蛋白质的亚基开放位点。
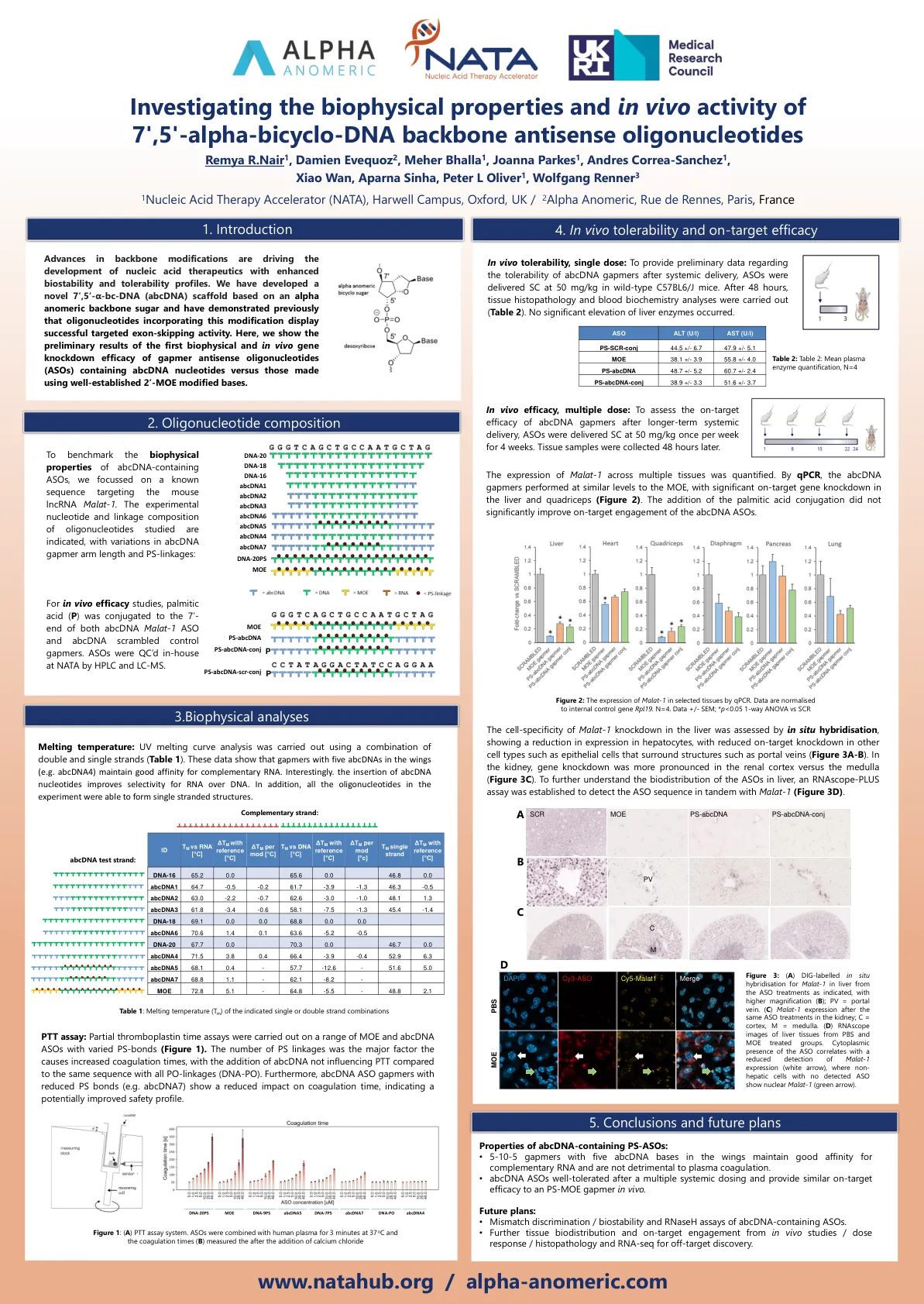
alpha-bicyclo-DNA骨架反义寡核苷酸
主链修饰的进步正在推动具有增强的生物稳定性和耐受性谱的核酸治疗剂的发展。我们已经开发了一种基于α异源主链糖的新型7',5'-α-BC-DNA(ABCDNA)支架,并先前证明了寡核苷酸含有这种修饰的寡核苷酸,该修饰显示了成功的靶向外显子鞋鞋。在这里,我们显示了含有AbcDNA核苷酸的Gapmer反义寡核苷酸(ASOS)的第一个生物物理和体内基因敲低功效的初步结果,而不是使用完善的2'MoE修饰碱基。

1 Genetic Testing: Whole Exome Sequencing and Whole Genome Sequencing
The human genome refers to an individual's complete set of DNA. The exome is a small section (1 to 2 percent) of the genome. It contains DNA sequences (exons) which provide instruction (coding) for making proteins, the building blocks of cells. Whole exome sequencing (WES) sequences only the coding region (1 to 2 percent) of an individual's genome. Whole exome sequencing can be used to identify variations in the protein-coding region of any gene rather than in only a select few genes. Because most known pathogenic variant(s) that cause disease occur in exons, WES is thought to be an efficient method to identify possible disease-causing pathogenic variant(s). Whole genome sequencing (WGS) sequences an individual's entire genome. It determines the order of all the nucleotides (the DNA building blocks) in an individual's DNA and can determine variations in any part of the genome. WES may potentially miss a pathogenic variant(s) in a non-coding region of the genome, therefore WGS may be used in selected cases if initial exome sequencing is not diagnostic. While WGS can accurately achieve copy number variation (CNV) detection, the use of chromosomal microarray analysis (CMA) continues to be the gold standard. Whole exome sequencing (WES) or whole genome sequencing (WGS) may be appropriate when there is no known cause of a patient's symptoms (e.g., prematurity, trauma, environmental, infectious, maternal immune disorder), clinical
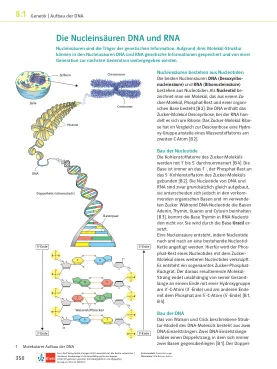
核酸DNA和RNA
核苷酸的构造糖分子的碳原子在1'至5英寸处编号[B 4]。碱始终与1'-,磷酸盐残基与糖分子的5´碳原子结合[B 2]。DNA和RNA的核苷酸通常是结构的,但是它们在前面的有机碱和糖的使用方面有所不同。虽然DNA-核苷酸含有腺苷,胸腺嘧啶,鸟嘌呤和胞嘧啶[B 3],但碱胸腺氨酸在RNA核酸中不发生。是由尿嘧啶基础制成的。核酸是通过逐渐将核苷酸添加到现有核苷酸链中而产生的。为此,核苷酸的磷酸盐其余部分与另一种核苷酸的糖分子有关。创建了所谓的糖磷酸骨链。所产生的分子链末端,无论其在一端的总长度如何,在3´-c原子(3´End)上的羟基和另一端,在5´-c原子(5´-end)上的磷酸盐[b 1,b 4]。

cas9 mRNA分离sec-mals
SepaxSRT®SEC1000Å和2000Å列在所有缓冲系统下的表现优于竞争对手的1000ÅSEC列,分辨率明显更高。sepax2000ÅSec柱在要筛选的三个列中的分辨率最高,这表明较大的孔径更适合Cas9 mRNA,具有4522个核苷酸尺寸。sepax srt sec表面化学还提供了独特的选择性和普遍的缓冲兼兼容性,其压力较低50%,这对于开发一种健壮,准确和可靠的分析方法很重要。

GO-CRISP 克隆协议
使用下面的引物模板(表 1),片段 2 可以通过 PCR 扩增(图 4A)。我们建议使用 gRNA NIA TLS1/2 作为 PCR 模板。“NNNNNNNNNNNNNNNNNNNN”(表 1)代表应由 gRNA 靶序列替换的核苷酸。用于创建 NIA1 靶向片段 2 的引物列于下方作为示例(表 1)——这些引物用于创建 gRNA NIA TLS1/2。引物还在片段末端添加了 BsaI 切割位点(图 4A),这些位点与 gRNA 片段 1 TLS1/2 中的双 BsaI 位点兼容。

非规范的 RNA-DNA 差异和其他人类基因组特征在非常短的串联重复中富集
非常短的串联重复序列在基因组分析中具有重要的遗传、进化和病理意义。本文,我们对 GRCh38 中的串联单核苷酸/二核苷酸/三核苷酸重复序列 (MNR/DNR/TNR) 进行了普查,我们统称其为“多束”。在人类基因组中,1.444 亿个核苷酸(4.7%)被多束占据,0.47 百万个单核苷酸被鉴定为多束铰链,即串联多束的断裂点。对普查的初步探索表明,AAC 多束的多束铰链位点和边界可能比其他多束区域具有更高的映射错误率。此外,我们揭示了近百种基因组特征的多束富集景观。我们发现 MNR、DNR 和 TNR 在杂项基因组特征(尤其是 RNA 编辑事件)的位置富集方面表现出明显差异。非规范和 C-to-U RNA 编辑事件在 MNR 内部和/或相邻处富集,而所有类别的 RNA 编辑事件在 DNR 中代表性不足。A-to-I RNA 编辑事件在多段中通常代表性不足。MNR 相邻范围内非规范 RNA 编辑事件的选择性富集为其真实性提供了负面证据。为了实现与多段相关的类似位置富集分析,我们开发了一个软件 Polytrap,它可以处理 11 个参考基因组。此外,我们将四种模式生物的多段编译成 Track Hub,它可以集成到 USCS Genome Browser 中作为官方轨道,以方便多段可视化。

DNA语言模型的核苷酸依赖性分析揭示了基因组功能元件
解密基因组中的核苷酸如何编码调节指令和分子机器是生物学的长期目标。DNA语言模型(LMS)通过对每个核苷酸的序列上下文进行建模概率来隐式捕获功能元素及其组织。但是,由于缺乏可解释的方法,使用DNA LMS发现功能基因组元素一直在挑战。在这里,我们引入了核苷酸依赖性,该核苷酸依赖性量化了一个基因组位置的核苷酸取代如何影响其他位置核苷酸的概率。我们生成了动物,真菌和细菌种类千倍体范围内成对核苷酸依赖性的全基因组图。我们表明,核苷酸依赖性比序列比对和DNA LM重建更有效地表明了人类遗传变异的有害性。调节元素在依赖图中显示为密集块,从而可以准确地对转录因子结合位点的系统识别,就像在实验结合数据上训练的模型一样准确地识别。核苷酸依赖性还突出了RNA结构内接触的基础,包括伪诺和三级结构接触,精确地。这导致发现了四个小说,实验验证的RNA结构中的大肠杆菌。最后,使用依赖图,我们通过基准测试和视觉诊断来揭示几种DNA LM体系结构和训练序列选择策略的临界局限性。完全,核苷酸依赖性分析为发现和研究功能元件及其在基因组中的相互作用开辟了新的途径。

DNA复制| HL IB生物学修订说明2025
如果腺嘌呤是原始链中的下一个暴露基础,则会添加胸腺核苷酸,如果胞嘧啶是原始链的下一个暴露基碱,则会添加鸟嘌呤核苷酸,并且仅根据与新的粘合物的规定,将鸟嘌呤核苷酸添加到新的基本链条之间,仅将基本构成形式添加到新的链接之间,如果构成的规则,则仅在水中构成了构成的规则。因此,新的DNA分子保留了一半的母体DNA,然后使用它来创建一个新的女儿链DNA复制在多细胞生物中很重要,原因是: