XiaoMi-AI文件搜索系统
World File Search Systemorbitals

第3章:原子中的电子
第3章:原子中的电子3.1亚壳和原子轨道3.2电子构造3.3电离能量学习结果:(a)描述主量子数量为1、2和3的S,P和D轨道的数量和相对能量,以及4S和4P Orbitals的S,P和D轨道。(b)描述S和P轨道的形状。(c)使用第1S²2S²2P⁶等质子数(和电荷)陈述原子和离子的电子配置等。(d)(i)解释并使用一词电离能。(ii)解释影响元素电离能的因素。(iii)解释了整个周期表的电离能量的趋势。(e)从连续的电离能量数据中推导元素的电子配置。(f)根据该元素在周期表中的位置来解释元素的连续电离能量数据。

在无限层镍模型中的抗磁相关性和近红色特征的出现
我们报告了一个由无限层镍元的启发的决定性量子蒙特卡洛研究,重点是层间杂交在3 d x 2-2-y 2轨道之间的影响,该杂交源自ni(或ni和o)在一个层中源自ni(或ni和o),在一个层和稀有(r)5 d orbitals in ni层中,ni and ni and and and and the ni and the and and and and and and and and and and libit。对于平均两层之间共有一个电子的填充,层间杂交会导致Ni层中的“自掺杂”孔,并且缺乏抗磁磁体排序,而是旋转密度和电荷密度条纹状状态的外观。随着层间杂交的增加,Ni和R层都会产生抗铁磁相关性,即使两个单独的层都远离半填充。用于中间范围内的杂交,大致可与内部的邻居跳跃跳跃t ni相提并论,该模型会形成近核样物理的特征。

在压力下,通过电子掺杂在三层镍中通过电子掺杂增强了S±波超导性的预测
是由最近报道的Trilayer LA 4 Ni 3 O 10在压力下的超导性的签名的动机,我们使用从头算和随机相近似技术全面研究了该系统。没有电子相互作用,NI D 3 z 2-r 2轨道显示通过op z轨道构成键合 - 抗抗反向和非键的分裂行为,这些轨道在La 4 Ni 3 O 10中诱导“ Trimer”晶格,类似于La 3 Ni 2 O 7的二聚体。费米表面由三个具有混合e轨道的电子纸组成,一个由D 3 z 2-r 2轨道组成的孔和电子袋组成,这表明Ni两轨最小模型。另外,我们发现由于M¼ðπ之间的部分嵌套,在S波通道中诱导了超导配对。以γ¼d为中心的费米表面的中心口袋和部分; 0Þ点。随着电子密度n的变化,S不稳定性保持领先,其配对强度显示出最大n¼4左右的Domelike行为。2(〜6。7%电子掺杂)。超导不稳定的消失在与新的1313堆叠La 3 ni 2 O 7中相同的电子密度消失,这与三层sublattice产生的孔口袋消失有关,这表明La 3 Ni 2 O 7的高t c超导性不源自trililayer and Monolayer and Monolayerererererereraner和monlayerererererereraner和mon。此外,我们在LA 4 ni 3 O 10中确认了拟议的自旋状态,其平面内(π,π)顺序(π,π)和底部Ni层之间的抗铁磁耦合,中间层中的旋转零。
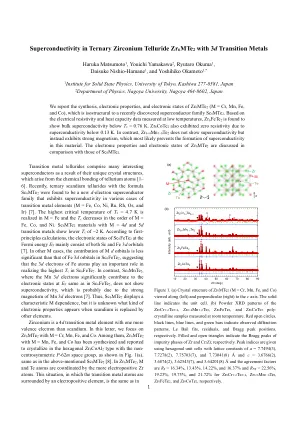
三元锆的超导性telluride zr6mte2 ...
过渡金属牙花会由于其独特的晶体结构而组成了许多有趣的超导体,这是由胎原子的化学键合引起的[1-6]。最近,发现带有配方SC 6 MTE 2的三元扫描库是一个新的D-电子超导体家族,在各种过渡金属元件的情况下表现出超导性(M = FE,CO,CO,NI,RU,RU,RH,RH,RH,OS和IR)[7]。在M = Fe中实现T C = 4.7 K的最高临界温度,而T C的M = Fe,Co和Ni的顺序下降。SC 6 MTE 2具有M = 4 d和5 d过渡金属的材料显示较低的t c 〜2 k。根据第一原理计算,Fermi Energy E F处的SC 6 Fete 2的电子状态主要由SC和Fe 3 D轨道组成[7]。在其他M情况下,M D轨道的贡献不如SC 6 FETE 2中的Fe 3 D轨道的贡献不那么重要,这表明Fe原子的3 d电子在实现SC 6 Fete 2中最高的T C中起着重要作用。相比之下,SC 6 MNTE 2,其中Mn 3 D电子在E f时与SC 6 Fete 2相同的电子状态显着促进了电子状态,并未显示超导性,这可能是由于Mn 3 D电子的强磁性引起的[7]。因此,SC 6 MTE 2显示了一个特征M的依赖性,但是当Scandium被其他元素取代时,尚不清楚出现哪种电子特性。

li8au电气中间质准原子状态的电子相关性和固有磁性
电气是一类不寻常的材料,其中间质阴离子电子(IAES)被捕获在带正电荷的晶格框架的有序腔中。与调用离子晶体相反,在电气中,仅由晶体中的原子轨道引起的占用能带(BRS)的占用能带的组合不应分解,但必要性应包括以电气位置为中心的准原子轨道的BR。1,限制在阴离子空位位置的此类电子的波函数表现出独特的双重性,结合了由动能与库仑相互作用之间的竞争引起的强烈定位和空间范围。这种竞争导致实现了复杂的多体基础状态。在某些情况下,原子和间质电子子系统之间的耦合非常弱,以至于可以单独考虑后者,从而为纯量子电子系统中现象的实现和研究创造了一个显着的平台。2,3,这种治疗

DNA 碱基的电子附着能
我们发现嘧啶胸腺嘧啶 (T) 和胞嘧啶 (C) 的 VAE 仅相差 0.03 eV,嘌呤鸟嘌呤 (G) 和腺嘌呤 (A) 的 VAE 仅相差 0.08 eV。与“化学”直觉相反,嘧啶的垂直形成的阴离子比较大嘌呤的阴离子更稳定,大约高 0.2 eV。考虑到每种化合物中中性势面和阴离子势面之间的 Franck-Condon 重叠,我们发现所有碱基都有一系列共同的能量,电子可在该能量范围内附着。换句话说,碱基的最低临时阴离子状态在实际意义上是简并的。此外,我们还观察到与腺嘌呤以外所有碱基的最低空分子轨道 (LUMO) 相关的临时阴离子核运动的证据。这表明电子注入这些轨道强烈激发中性分子的振动模式。

Crystalline电池材料中的离子迁移率
图2:(a)MRSF-TDDFT计算的工作流程,(b)分支分为三个不同的MRSF-TDDFT计算,具有从相同的KS轨道衍生的不同自旋状态,以及(c)基于两个不同电子状态的梯度计算,分支为同一MRSF-TDDDDFT FTDFT ENCELINC,所有使用MRSF-TDDDDFT FTD FTD FENT态,所有利用了Pyty-accce python-accce python-accce cess。

分子轨道洞察等离子体诱导的甲烷光解
摘要:等离子体诱导光催化是一种降低传统热分解温度的有效方法,已被用于甲烷脱氢。本文,我们利用时间相关密度泛函理论,通过分子轨道洞察,探讨了等离子体诱导甲烷在四面体 Ag 20 纳米粒子上解离的微观动力学机制。我们巧妙地通过 Hellmann-Feynman 力建立了化学键和分子轨道之间的关系。时间和能量分辨的光载流子分析表明,由于 Ag 纳米粒子和 CH 4 轨道的强杂化,在低激光强度下,从 Ag 纳米粒子到甲烷的间接热空穴转移主导光反应,而间接和直接电荷转移共存,促进甲烷在强激光场中的解离。我们的研究结果可用于设计新型甲烷光催化剂,并强调了分子轨道方法在吸附质-底物体系中的广阔前景。关键词:局域表面等离子体、甲烷脱氢、光载流子动力学、分子轨道洞察、实时时间相关密度泛函理论

在滑动铁电
先前的实验提供了分别在二维材料中滑动铁电性和光激发层间剪切位移的证据。在这里,我们发现通过激光照明,在H -BN双层中令人惊讶的0.5 ps中可以实现垂直铁电的完全逆转。综合分析表明,铁电偏振转换源自激光诱导的层间滑动,这是由多个声子的选择性激发触发的。从上层n原子的P z轨道到下层B原子的P z轨道的层间电子激发产生所需的方向性层间力,激活了平面内光学TOTO TOTO TOS TOTO to-1和LO-1声音声模式。由TO-1和LO-1模式的耦合驱动的原子运动与铁电软模式相干,从而调节了动态势能表面并导致超快铁电偏振反转。我们的工作为滑动铁电的超快偏振转换提供了一种新颖的微观见解。

超低氧气氛中半导体氧化钇薄膜的反应脉冲直流磁控溅射沉积:光谱
摘要 利用反应脉冲直流磁控溅射技术进行了一项实验研究,探索了在 623 K (± 5K) 下沉积的半导体氧化钇薄膜的光谱和结构特性。根据 x 射线衍射和透射电子显微镜测量的结果,一氧化钇很可能在 β-Y 2 O 3 和 α-Y 2 O 3 之间的过渡区中形成,并伴有晶体 Y 2 O 3 。由于 4d 和 5s 轨道之间的能量分离低和/或相应轨道亚能级的自旋状态不同,一氧化物的稳定性在热力学意义上最有可能受晶体大小的自身限制。与金属氧化物立方结构相比,这种行为会导致晶体结构扭曲,并且还会影响纳米晶/非晶相的排列。此外,椭圆偏振光谱法表明半导体氧化钇的形成特征比结晶的 Y 2 O 3 更显著,且大多为非晶态。我们的目的是利用目前的研究结果,加深对不寻常价态 (2+) 钇的形成动力学/条件的理解。