XiaoMi-AI文件搜索系统
World File Search Systemsapiens

重新设定
甲虫巨星Jannaschi(古细菌)1.7 100-200 X / 0.1-0.2 E. Coli K12(细菌)4.6 100-200× / 0.5-0.5-0.5-0.5-0-0。 1-1.2秀丽隐杆线虫(线虫蠕虫)97 80-100-100× / 8-10拟南芥(植物)125 80-100-100× / 10-13果蝇黑色素果(果蝇)180 80-100-100-100× / 15-18 Danio Rerio(斑马鱼)1400 30-50× / 42-70 HOMO SAPIENS(HUMAN)3300 30× / 99(浅层); ≥80×/≥264(DEP)Hordeum dufgarre(大麦)4200 30× / 126 BUFO BUFO(TOAD)5000 30× / 150× / 150× /

白色念珠菌的计算机代谢途径分析……
白色念珠菌是一种真菌或酵母,通常生长在口腔和消化道中,可引起念珠菌病。白色念珠菌可从良性共生菌转变为致病病原体,导致口腔、胃肠道和生殖道感染。复发性阴道鹅口疮是由对念珠菌病的局部免疫缺陷引起的,这可能是由于前列腺素 E2 (PGE2) 合成过多引起的。白色念珠菌菌株的致病性存在差异,这表明菌株特有的毒力因子可能在疾病严重程度中发挥作用。对宿主智人和病原体白色念珠菌的代谢途径进行了计算机比较分析。blastp e 值阈值截止值设置为 0.005。 281 种酶中,共有 118 种酶序列与人类蛋白质序列非同源,其中 24 种酶根据 DEG 数据库被发现对白色念珠菌的生存至关重要。CELLO v.2.5:subCELlulor 定位预测器结果显示,约 57% 的酶位于细胞质中,15% 的酶位于线粒体中,12% 的酶为质膜蛋白,6% 的酶位于细胞核中,5% 的酶位于叶绿体和过氧化物酶体中。确定的潜在药物靶点为进一步研究发现新型治疗化合物奠定了基础。
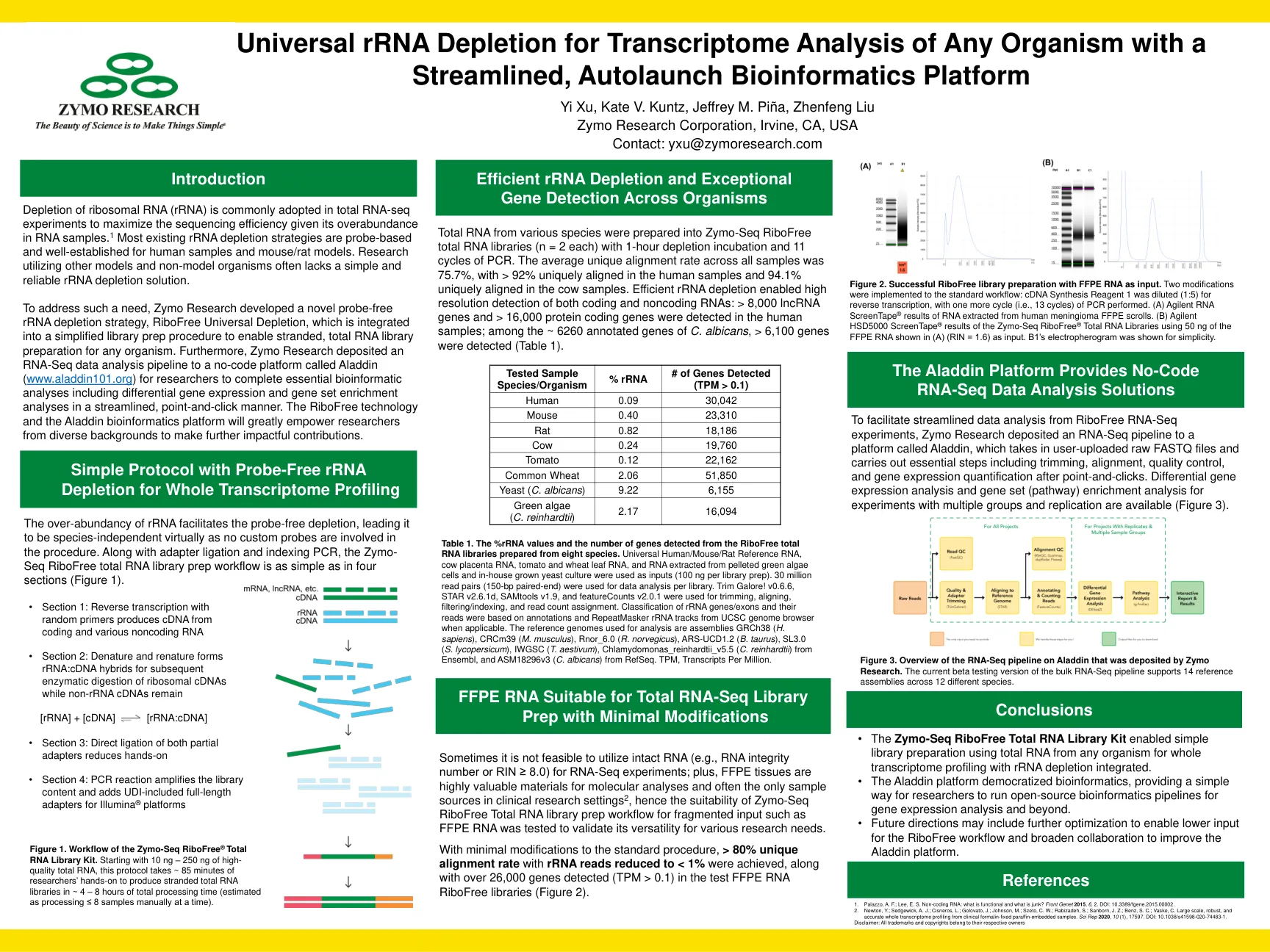
通用rRNA耗竭用于任何有机体的转录组分析
表1。从八个物种制备的无核能总RNA文库中检测到的%rRNA值和基因数量。通用的人/小鼠/大鼠参考RNA,牛胎盘RNA,番茄和小麦叶RNA以及从沉淀的绿藻细胞中提取的RNA和内部成年酵母菌培养物用作输入(每位图书馆100 ng)。每个库的数据分析使用了3000万读对(150 bp配对)。修剪大奖!v0.6.6,Star v2.6.1d,Samtools v1.9和farmaturecounts v2.0.1用于修剪,对齐,过滤/索引和读取计数分配。RRNA基因/外显子的分类及其读取是基于UCSC基因组浏览器的注释和retoMasker rRNA轨道的基础。 用于分析的参考基因组是组件GRCH38(H。SAPIENS),CRCM39(M。MUSCULUS),RNOR_6.0(R。NORVEGICUS),ARS-UCD1.2(B. Taurus),SL3.0,SL3.0,SL3.0(S. lycopersicum),IWGSC(iwgsc),IWGSC(iwgsc),iwgsc(iwgscim),chlamans,C。c. c. c. c. c. c. c. anasen nasunson。 Reinhardtii)来自Ensembl和Refseq的ASM18296V3(C. albicans)。 tpm,百万分的成绩单。RRNA基因/外显子的分类及其读取是基于UCSC基因组浏览器的注释和retoMasker rRNA轨道的基础。用于分析的参考基因组是组件GRCH38(H。SAPIENS),CRCM39(M。MUSCULUS),RNOR_6.0(R。NORVEGICUS),ARS-UCD1.2(B. Taurus),SL3.0,SL3.0,SL3.0(S. lycopersicum),IWGSC(iwgsc),IWGSC(iwgsc),iwgsc(iwgscim),chlamans,C。c. c. c. c. c. c. c. anasen nasunson。 Reinhardtii)来自Ensembl和Refseq的ASM18296V3(C. albicans)。tpm,百万分的成绩单。
预定的教学活动/预期教学
docente:克里斯蒂安·卡皮利(Cristian Capelli)教授的化石遗骸分子分析(“古代DNA”)代表了近年来引起了浓厚兴趣的研究领域之一,不仅在该领域的专业人员中。可以重建过去有机体的DNA的想法无疑具有超越科学期刊页面的魅力,并轻松吸引了公众的注意。在本课程中,我们将探讨什么是古老的DNA以及如何从考古,历史和博物馆遗体中回收的生物材料。我们将检查这种方法的局限性和潜力,并对塑造其发展的事件进行积极和负面的构成。最后,我们将分析一些最重要的结果,尤其是那些与理解我们物种的进化史 *HOMO SAPIENS *相关的结果。

与人工智能相关的人格:法律问题
技术的作用至关重要,并且一直在不断发展,使人类的生活更加轻松。世界目前正在经历第四次革命,这场革命以人工智能为基础。人工智能 (AI) 是在 20 世纪 50 年代末作为专家系统研究的一部分而开发的。这项研究基于这样的信念:如果人类可以解决常识问题,那么机器也可以。它试图用人工智能技术取代人类的能力。人工智能在各个领域的应用越来越多,这可以归因于其提高生产力和确保快速有效的解决问题的能力。该领域研究的顶峰是开发了一种真正的思考机器,称为“Machina Sapiens”,它可以像人类一样行动和推理。1 然而,这些特征被发现不足以实现智能思考。因此,添加了更多属性,例如沟通、内部知识、外部知识、目标驱动行为和创造力。2

物理学,觅食流动性和Sahul的第一个人
Homo Sapiens进入Sahul的路线和速度仍然是考古学的主要研究问题。在这里,我们介绍了一种方法,该方法通过将随时间不断发展的景观与Lévy步行觅食模式结合起来,对人类流动性的影响进行建模,后者构成了短途步骤的结合以及偶尔的更长的移动,而猎人 - 总成员可能会用于新环境的效率探索。我们的结果表明,在河道走廊和海岸线之后,遍布萨哈尔的散布浪潮。基于考古遗址和预测的距离估计迁移速度属于先前报道的Sahul和其他地区的范围。从我们的机械移动模拟中,我们分析了考古遗址的可能性,并突出了澳大利亚具有考古潜力的地区。我们的方法补充了现有的方法,并提供了有关萨哈尔更新世考古学的有趣观点,这些观点可以应用于世界其他地区。

dnabert,一种在生物学和健康中进行顺序预测的语言方法
本文论文讨论了这种新的DNABERT模型,并解决了它对生物学和健康产生影响的程度。在这里,与当前现有模型相比,DNABERT是否是革命性的。通过比较先前研究中预测模型的准确性与DNABERT的准确性,我得出的结论是,DNABERT可以在剪接位点预测上获得出色的性能,并且可以获得最高的准确性,但无法获得启动子预测的出色性能。因此,我的目的是确定DNABERT的工作原理,以便可以获得可能可以用于进一步优化和自定义的理解。因此,分析了DNABERT的K-MER令牌化方法和字节对编码。这是通过采用Ji等人的DNABERT的所述方法来进行的。(2021)和Zhou等人的DNABERT-2。(2023)。从此分析中可以得出结论,两种方法都比现有的DNA/RNA预测方法更好,但是BPE是最有前途的。之后,使用DNABERT(DNABERT-PROM)重点介绍了启动子预测,以清楚地了解其过程以及如何进行预培训。为了获得此信息,Ji等人的DNABERT-PROM方法的描述。(2021)进行了调整。在这里,可以确定的是,使用具有TATA-Box存在或不存在的远端启动子,对DNABERT-PROM进行了培训,以预测Homo Sapiens。此外,使用EPDNEW数据库获取启动子的数据。为此,Ji等人的DNABERT的描述特性。在分析了DNABERT-PROM之后,我得出的结论是,它是一个高效的模型,可以预测Homo Sapiens中的启动子。最后,我选择提供更广泛的DNABERT观点,以研究如何在生物学和健康领域中应用。(2021)进行了调整,并将其与生物学和健康中的当前限制进行了比较。在这里,我得出的结论是,DNABERT是生物学和健康中转录调节预测的最有前途的模型,因为它可以解决上下文所需的信息。我得出的结论是,DNABERT也应该是执行其他类型的DNA/RNA预测的“第一选择”方法,尽管它们的用法绝不能替代研究和诊断中的决策。尽管DNABERT已经是一个非常充分的预测模型,但仍需要进一步的优化和自定义来扩大其对生物学和健康中顺序预测的贡献。

超越有限理性 - eScholarship
错误的决策可能会带来灾难性的后果,大量文献表明,人类的判断和决策充斥着大量违反逻辑、概率论和预期效用理论规则的系统性行为。20 世纪 70 年代发现这些认知偏见,挑战了智人作为理性动物的概念,并深刻动摇了认知、神经和社会科学中经济学和理性模型的基础。四十年后,这些学科仍然缺乏能够解释人们认知偏见的严格理论基础。此外,设计有效的干预措施来纠正认知偏见并改善人类的判断和决策仍然是一门艺术,而不是一门科学。我在论文的第一部分和第二部分分别讨论了这两个基本问题。

超级金属:一种生成的AI框架,用于蛋白质中的快速,精确的金属离子位置预测
最初的计算方法用于mRNA定位是单个标签分类任务,其中每个mRNA被预测仅定位为一个特定的隔室。rnatracker采用了深层复发的神经网络来预测mRNA定位[6]。iloc-mRNA,利用支持载体机(SVM)来预测在同性恋中的mRNA定位,[7]。sublocep通过集中在特定的细胞室,同时保留在单标签分类框架内[8],进一步完善了预测。但是,它们本质上受到这样的假设,即每个mRNA仅定位到一个与生物学现实不符的隔间。许多mRNA众所周知,可以定位在多个隔间中,从而在细胞内履行各种作用[9,8]。

光转导基因和感光细胞的前脑前诊所起源
图1。光转传成分基因家族和真核生物中的分布的进化史。重建了所有常见(a),横幅特异性(b)和睫状特异性(c)成分的每个基因家族的演变(每个部分顶部的基因树),并且它们的分布均映射在eukarya的主要群体中(每个部分左侧的物种树)。的存在用一个完全彩色的圆表示。在每个基因家族中,含有D. melanogaster(红色),H。sapiens(绿色)或(蓝色)基因(S)在光转传途径中起作用的(蓝色)基因(S)的感兴趣的亚家族。在几个基因家族中,根据系统发育,某些注释较差的序列与感兴趣的群体非常遥远。这些进化枝被标记为“不确定”。实际上,它们可以代表基因家族的真正相关成员,因为它们是在数据挖掘过程中检索并在管道期间保留的。但是,不能排除他们宁愿属于另一个基因家族。