XiaoMi-AI文件搜索系统
World File Search Systemsequences

DNA 超声逆向 PCR 用于基因组规模分析未知侧翼序列
图 1 超声逆向 PCR (SIP) 的可视化表示。图中使用的缩写包括 KoRV — 考拉逆转录病毒、LTR — 长末端重复、pol — 聚合酶基因。 (a) 整合到考拉基因组 DNA 中的 KoRV 原病毒以典型的 LTR 区域 (绿色框) 和逆转录病毒基因 (蓝色框) 两侧的形式显示。注意:为简单起见,仅以图表形式表示 pol 基因 (红色框) 的大致位置。 (b) 使用超声处理将考拉基因组 DNA 碎裂成平均长度为 2-7 kb 的片段。然后对碎裂的 DNA 进行平端修复和磷酸化 (未显示)。 (c) 随后将样品分成两部分:非适配器组 (c1) 和适配器组 (c2)。非接头组在环化之前未进行任何修改,而接头组在 DNA 分子的两端连接有相同的接头序列(黄色框),用于辅助解释环化和扩增后的倒置扩增子序列。(d)接头组和非接头组均环化,从而产生环状 DNA 模板。(e)环状 DNA 模板用两组针对 KoRV 的 pol 和 LTR 区域的引物进行扩增。没有这些引物结合位点的环状模板不会扩增。(f)扩增和测序产物被倒置,引物结合位点位于扩增子的侧翼。产生了两种主要类型的 PCR 产物:(i)由 LTR 引物扩增的 PCR 产物和(ii)由 pol 引物扩增的 PCR 产物

使用自由能量优化的互补工作记忆来学习序列中的特征和结构
摘要 — 我们提出了一个整体框架,用于对皮质基底系统 (CX-BG) 和额叶纹状体系统 (PFC-BG) 进行建模,以生成和回忆音频记忆序列;即声音感知和语音产生。我们真正的模型基于称为 INFERNO 的神经结构,代表循环神经网络的迭代自由能优化。自由能 (FE) 对应于内部或外部噪声的预测误差。FE 最小化用于在 PFC 中探索、选择和学习在 BG 网络中执行的最佳操作选择(例如声音产生),以便尽可能准确地重现和控制代表 CX 中声音的脉冲序列。两种工作记忆之间的差异依赖于神经编码本身,它基于 CX-BG 网络中的时间排序(脉冲时间依赖可塑性)和 PFC-BG 网络中序列的排序(门控或增益调制)。我们在这篇短文中详细介绍了负责以几毫秒的顺序对音频基元进行编码的 CX-BG 系统,以及负责学习序列中时间结构的 PFC-BG 系统。使用小型和大型音频数据库进行的两个实验展示了神经架构在检索音频基元以及基于结构检测的长距离序列方面的探索、泛化和抗噪能力。虽然两种学习机制都是用相同的顺序编码算法实现的,但 CX-BG 系统实现了无模型循环神经网络 (INFERNO),而 PFC-BG 系统实现了门控循环神经网络 (INFERNO GATE)。

使用 transformers 根据 DNA 序列和转录后信息预测基因表达水平
背景与目标:近年来,由于基因表达水平的潜在临床应用,预测基因表达水平至关重要。在此背景下,Xpresso 和其他基于卷积神经网络和 Transformer 的方法首次被提出用于此目的。然而,所有这些方法都使用标准的独热编码算法嵌入数据,从而产生非常稀疏的矩阵。此外,该模型没有考虑基因表达过程中最重要的转录后调控过程。方法:本文提出了 Transformer DeepLncLoc,一种通过处理基因启动子序列来预测 mRNA 丰度(即基因表达水平)的新方法,将该问题作为回归任务进行管理。该模型利用基于 Transformer 的架构,引入 DeepLncLoc 方法执行数据嵌入。由于 DeepLncloc 基于 word2vec 算法,因此它避免了稀疏矩阵问题。结果:该模型包含了与 mRNA 稳定性和转录因子相关的转录后信息,与最先进的方法相比,其性能显著提高。Transformer DeepLncLoc 的 R 2 评估指标达到 0.76,而 Xpresso 的 R 2 评估指标为 0.74。结论:Transformer 方法中的多头注意力机制适用于对 DNA 位置之间的相互作用进行建模,从而克服了循环模型。最后,在管道中整合转录因子数据可显著提高预测能力。


变压器模型产生的噬菌体基因组在组成上与自然序列不同
语言模型在基因组学中的新应用有望对该领域产生重大影响。Megadna模型是创建合成病毒基因组的第一个公开可用的一代模型。评估Megadna概括病毒的非随机基因组组成以及是否可以通过算法检测到合成基因组,4,969个天然噬菌体基因组和1,002 de Novo合成细菌噬菌体的组成指标比较了。变压器生成的序列已通过Genomad分类为变化但现实的基因组长度,而58%的序列分类为病毒。然而,与天然的Bacte-riophage基因组相比,通过秩-SUM测试和原理分析分析,这些序列在各种综合度量中呈现一致的差异。一个简单的神经网络训练,可在全球组成指标上检测变压器生成的序列,其中位灵敏度为93.0%,特异性景观为97.9%(n = 12个独立模型)。总体而言,这些恢复表明,巨型群岛尚未具有逼真的组成偏见,并且基因组组成是检测该模型产生的序列的可靠方法。虽然结果是Megadna模型的特异性,但此处描述的评估框架可以应用于基因组序列的任何生成模型。

通过选择性去除冗余核酸序列
metatranscriptome(metat)测序是分析微生物组动态代谢功能的关键工具。除了分类信息外,Metat还提供了宿主和微生物种群的实时基因表达数据,从而允许对微生物组及其宿主的功能(酶)输出的真实定量。有效且准确的元数据分析的主要挑战是从这些复杂的微生物混合物中去除高度丰富的rRNA转录本,这些混合物可以在数千个种类中进行数量。不管rRNA耗竭的方法论如何,基于微生物组的分类学含量的RRNA去除探针的设计通常需要大量的单个探针,这使得这种方法使商业上生产,昂贵且经常在技术上不可行。在先前的工作[1]中,我们使用仅基于序列丰度的设计策略为人类粪便样品设计了一组耗竭探针,完全不可知的是存在的微生物物种。在这里,我们表明,与小鼠盲肠样品一起使用时,基于人类的探针效果较差。然而,将其他rRNA耗竭探针专门针对盲肠含量提供了更高的效率和一致性,以用于对小鼠样品的元分析。

DNA中DGPDC和DCPDG序列的光活化动力学:全面的量子机械研究
通过DNA吸收紫外线是细胞氧化损伤的主要来源,引发了一系列对生物体的可能非常有害结果的分子事件(DNA突变,凋亡和癌症)。1 - 3,因此,巨大的效果已致力于表征多核苷酸的光活化动力学。归功于时间分辨(TR)光谱技术4 - 6的发展以及量子机械(QM)计算的限制,已经取得了7 - 10个重要的进步,尤其是在模型多核苷酸序列的研究中。7 - 9,11 - 13他们的光活化动力学非常复杂,结合了超高过程,其特征是亚匹克秒(PS)中的时间常数多达几个PS,而其他过程则以较低的时间尺度出现,最高为纳米秒(NS)(NS)及以后。最快的过程通常与单体样衰减过程有关,即类似于孤立基地中发生的,而,而

表Si.定量RT-PCR的序列序列。
a,siRNA名称/方向序列atp6v1b1_#1 sense 5' - gacaacuucgccaucgucu-3'反义5'-agacgauggcgaaguugu -3'atp6v1b1_#2 ′ B, qPCR primers Gene/direction Sequence ATP6V1B1 Fw 5 ′ -CAGCAGGCTCAGACACTGG-3 ′ Rev 5 ′ -CCCAGGCCTGCTGTCTATCTC-3 ′ Cyclin D1 Fw 5 ′ -CCGTCCATGCGGAAGATC-3 ′ Rev 5 ′ -ATGGCCAGCGGGAAGAC-3 ′ p21 Fw 5 ′ -AGTCAGTTCCTTGTGGAGCC-3 ′ Rev 5 ′ -CATTAGCGCATCACAGTCGC-3 ′ GAPDH Fw 5 ′ -AGAAGGCTGGGGCTCATTTG-3 ′ Rev 5 ′ -AGGGGCCATCCACAGTCTTC-3 ′ AccuTarget Negative Control siRNAs, catalogue no.SN-1013(Bioneer Corporation)。fw,前进; REV,反向; siRNA,小干扰RNA; ATP6V1B1,ATPase H+运输V1亚基B1。

用感知器的DNA和蛋白质序列预测基因和蛋白质表达水平
背景和客观:生物体的功能及其生物学过程源于基因和蛋白质的表现。因此,量化和预测mRNA和蛋白质水平是科学研究的关键方面。关于mRNA水平的预测,可用的方法使用转录起始位点(TSS)上游和下游的序列作为神经网络的输入。最新模型(例如Xpresso和basenjii)预测利用卷积(CNN)或长期记忆(LSTM)网络的mRNA水平。但是,CNN预测取决于卷积内核的大小,LSTM遭受捕获序列中的长期依赖性。据我们所知,关于蛋白质水平的预测,没有通过利用基因或蛋白质序列来预测蛋白质水平的模型。方法:在这里,我们利用一种新的模型类型(称为感知器)用于mRNA和蛋白质水平预测,从而利用了具有注意力调节的基于变压器的体系结构来参加序列中的长期相互作用。此外,感知器模型克服了标准变压器体系结构的二次复杂性。这项工作的贡献是1。dnaper-ceiver模型,以预测TSS上游和下游序列的mRNA水平; 2。Pro-teminepeiver模型,以预测蛋白质序列的蛋白质水平; 3。蛋白质和dnapceiver模型,以预测TSS和蛋白质序列的蛋白质水平。结果:这些模型是在细胞系,小鼠,胶质母细胞瘤和肺癌组织上评估的。结果表明,感知器型模量在预测mRNA和蛋白质水平方面的有效性。结论:本文介绍了mRNA和蛋白质水平预测的感知器结构。将来,将调节和表观遗传信息插入模型可以改善mRNA和蛋白质水平的预测。源代码可在https://github.com/matteostefanini/dnaperceiver
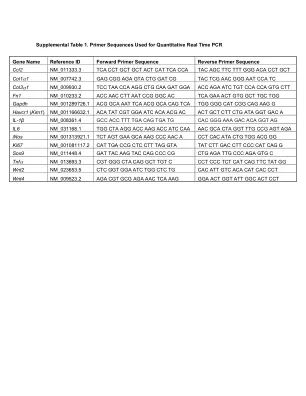
补充表1。用于定量实时PCR的引物序列
havcr1(kim1)NM_001166632.1 ACA TAT CGT GGA ATC ACA ACG ACG AC AC AC AC ACT GCT CTT CTG CTG ATA GGT GAC A