XiaoMi-AI文件搜索系统
World File Search Systemwhole

将飞机电气化提升到一个全新的水平
挤压、胶带包裹和屏蔽胶带包裹电缆 我们所有的高 1000 V 电缆均采用高度灵活的镀镍铜线制成,规格从 #8 到 #0000 AWG,适用于苛刻的飞行剖面和高达 260°C 的温度。这些电缆共同解决了 EWIS 工程师在设计配电系统时遇到的常见问题。挤压电缆具有出色的可剥离性;复合电缆具有抗磨损、直径小、重量轻的特点;屏蔽电缆具有出色的 EMI 控制和故障检测能力。所有三个产品系列均可激光标记,以便于识别。

HPV E6/E7 mRNA测定,HPV DNA测试和细胞学的疗效在检测高级宫颈病变和侵入性癌症中的疗效
Joseph Agofure Idogho剧院和媒体艺术系,联邦大学Oye Ekiti,Ekiti州,尼日利亚Ekiti State,摘要唱歌和音乐,这是艺术 /戏剧的重要方面,在每种文化中都起着重要作用;特别是在儿童发展中。音乐无疑是我们生活,电视,电影,宗教崇拜,庆祝活动等的许多方面存在的。从出生开始,父母本能地利用音乐来平静/舒缓孩子,表达他们的爱与喜悦,互动和互动。但是,对儿童的整体发展和这种艺术一样重要;在尼日利亚的儿童护理人员和幼儿园的老师中,它不足。本研究记录了尼日利亚Oye Lga Ekiti的幼儿园老师和学生的看法,内容涉及在课堂上通过音乐/歌曲学习的进口。焦点小组讨论与48位教师和30(30)学生(4-6岁)进行了讨论。参与者被要求他们对音乐/歌曲作为课堂教学策略的了解和态度。不断的比较分析或方法表明,教师对音乐/歌曲在课堂上的作用有很好的了解。但是,诸如完成教学大纲的压力之类的因素,担心在学生/同事面前失去尊重以及它产生的噪音抑制了教师在教室中使用音乐/歌曲的使用。学生承认他们喜欢通过音乐/歌曲学习,因为它可以帮助他们轻松吸收。调查结果表明,音乐/歌曲是整个儿童发展的名副其实的工具,尽管严格的学术计划会影响其在课堂教学学习环境中的接受。因此,这项研究表明,政策制定者和政府必须培训教师在教学过程中有意利用音乐 /歌曲,尤其是在幼儿教育环境中。关键字:音乐,年龄段,教育,整个孩子,发展

2024-2025年整个学校课程和评估计划。...
▪ 优先考虑每个学生学习历程中的关键点 ▪ 关注教育成就、幸福感和参与度以及文化和包容性。麦凯北州立高中在考虑政府优先事项并与家长和学校社区协商后实施 P-12 框架的要求。我们通过有效的学校领导、优质的教学和学习来满足要求,注重改进系统的课程交付,以确保所有身份和能力的学生都能够: • 与同龄人一起学习和参与课程 • 通过量身定制的支持(包括满足其学习需求的合理调整)在学业和社交方面取得成就。
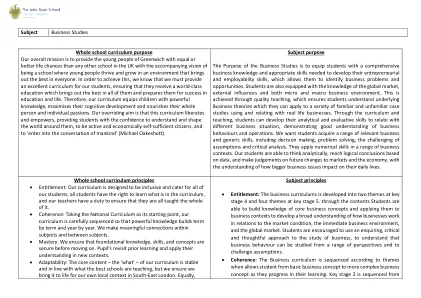
主题商业研究整个学校课程...
整个学校课程的目的我们的整体任务是为格林威治的年轻人提供相同或更好的生活机会,而英国的任何其他学校的生活机会都具有伴随的愿景,即成为一所年轻人在一个使每个人中最好的环境中成长和成长的学校。为了实现这一目标,我们知道我们必须为我们的学生提供出色的课程,并确保他们接受世界一流的教育,这使他们在所有人中都提供了最好的教育,并为他们的教育和生活取得成功做好了准备。因此,我们的课程使儿童具有强大的知识,最大程度地提高了他们的认知发展,并滋养了他们的整个人和个人激情。我们的压倒性目标是,该课程解放和授权者,使学生有信心理解和塑造周围的世界,积极且经济自给自足的公民,并“进入人类的对话”(Michael Oakeshott)。

全外显子组测序和 CRISPR/ 的联合使用...
1 法国格勒诺布尔阿尔卑斯大学先进生物科学研究所、INSERM U1209、CNRS UMR 5309、38000 格勒诺布尔、不孕症遗传学表观遗传学和治疗团队; EXT-CCazin@chu-grenoble.fr(抄送); corinne.loeuillet@univ-grenoble-alpes.fr (法语); christophe.arnoult@univ-grenoble-alpes.fr(加拿大); PRay@chu-grenoble.fr (PFR) 2 UM GI-DPI, CHU Grenoble Alpes, 38000 格勒诺布尔, 法国; ilordey@chu-grenoble.fr 3 日内瓦大学医学院遗传医学与发展系,CH-1211 Genève 4,瑞士; Yasmine.NEIRIJNCK@univ-cotedazur.fr(YN); Lydia.Wehrli@unige.ch (LW); Francoise.Kuhne@unige.ch (FK); Serge.Nef@unige.ch (SN) 4 突尼斯医疗援助中心,Polyclinique les Jasmins,Centre Urbain Nord,突尼斯 1003; fourati_selima@yahoo.fr(SFBM); aminbouker@gmail.com (AB); raoudha.zouari@cliniquelesjasmins.com.tn (RZ) 5 TIMC-IMAG,CNRS 和格勒诺布尔阿尔卑斯大学,38000 格勒诺布尔,法国; Nicolas.Thierry-Mieg@univ-grenoble-alpes.fr * 通信地址:ZEKherraf@chu-grenoble.fr;电话:+33-(0)4-7676-8303

全外显子组测序和 CRISPR 的联合使用...
1 法国格勒诺布尔阿尔卑斯大学先进生物科学研究所、INSERM U1209、CNRS UMR 5309、38000 格勒诺布尔、不孕症遗传学表观遗传学和治疗团队; EXT-CCazin@chu-grenoble.fr(抄送); corinne.loeuillet@univ-grenoble-alpes.fr (法语); christophe.arnoult@univ-grenoble-alpes.fr(加拿大); PRay@chu-grenoble.fr (PFR) 2 UM GI-DPI, CHU Grenoble Alpes, 38000 格勒诺布尔, 法国; ilordey@chu-grenoble.fr 3 日内瓦大学医学院遗传医学与发展系,CH-1211 Genève 4,瑞士; Yasmine.NEIRIJNCK@univ-cotedazur.fr(YN); Lydia.Wehrli@unige.ch (LW); Francoise.Kuhne@unige.ch (FK); Serge.Nef@unige.ch (SN) 4 突尼斯医疗援助中心,Polyclinique les Jasmins,Centre Urbain Nord,突尼斯 1003; fourati_selima@yahoo.fr(SFBM); aminbouker@gmail.com (AB); raoudha.zouari@cliniquelesjasmins.com.tn (RZ) 5 TIMC-IMAG,CNRS 和格勒诺布尔阿尔卑斯大学,38000 格勒诺布尔,法国; Nicolas.Thierry-Mieg@univ-grenoble-alpes.fr * 通信地址:ZEKherraf@chu-grenoble.fr;电话:+33-(0)4-7676-8303

Cas9 的组织特异性表达对整体没有影响......
目标:CRISPR/Cas9 技术彻底改变了基因编辑,并加快了我们操纵目标基因的能力,以造福研究和治疗应用。尽管该领域已经取得了许多进展,并且还在继续取得进展,但迄今为止使用最广泛的技术可能是产生敲除细胞、组织和动物。该技术的优势是多方面的,但关于长期表达 Cas9 等外来蛋白质对哺乳动物细胞功能的影响,仍然存在一些问题。几项研究表明,Cas9 的慢性过度表达(无论是否伴有其伴随的向导 RNA)可能会对细胞功能和健康产生有害影响。在体内应用该技术时,这一点尤其令人担忧,因为 Cas9 在目标组织中的慢性表达可能会促进疾病样表型,从而混淆对目标基因影响的研究。尽管这些担忧仍然存在,但据我们所知,尚无任何研究直接证明这一点。方法:在本研究中,我们使用 lox-stop-lox (LSL) spCas9 ROSA26 转基因 (Tg) 小鼠系生成了四种组织特异性 Cas9-Tg 模型,这些模型在心脏、肝脏、骨骼肌或脂肪组织中表达 Cas9。我们对这些小鼠进行了全面的表型分析,直至 20 周龄,随后对其器官进行了分子分析。结果:我们证明 Cas9 在这些组织中的表达对动物的全身健康没有不利影响,也不会对全身能量代谢、肝脏健康、炎症、纤维化、心脏功能或肌肉质量产生任何组织特异性影响。结论:我们的数据表明,这些模型适用于使用 LSL-Cas9-Tg 模型研究基因缺失的组织特异性效应,并且利用这些模型观察到的表型可以自信地解释为基因特异性的,并且不会因 Cas9 的慢性过表达而混淆。2021 作者。由 Elsevier GmbH 出版。这是一篇根据 CC BY-NC-ND 许可协议开放获取的文章(http://creativecommons.org/licenses/by-nc-nd/4.0/)。

整个病毒体,灭活冠状病毒疫苗
所有制剂均在小鼠、大鼠和兔子身上进行了免疫原性测试。小鼠、大鼠和兔子在第 0、7 和 14 天(n+1 剂量)接种疫苗。此外,在叙利亚仓鼠攻击模型和非人类灵长类动物(恒河猴)攻击模型中测试了这些制剂的免疫原性、安全性和保护功效。仓鼠在第 0、14 和 35 天(n+1 剂量)接种疫苗,活 SARS-CoV-2 病毒在第 50 天通过鼻内途径进行攻击。同样,恒河猴在第 0 天和第 14 天接种疫苗,活 SARS-CoV-2 病毒在第 28 天通过鼻内和气管内途径进行攻击。所有制剂均被发现是安全的、免疫原性的,并为上呼吸道和下呼吸道提供有效保护。 6. 药物详情 6.1 辅料清单

6. 2023-24 年全校战略计划
受托人监测 RAG ● 每学期审查战略计划 ● 每学期校长报告 ● 匿名 TOT ● 匿名 Perf 管理计划 ● ISP 数据 ● 领导与管理链接访问 ● 顾问访问记录 ● 进行并分享员工调查 实施 - 我们将要做什么 谁 何时 资源 ARU CPD 发生以设置和支持流程 - 员工会议和培训日 EC 秋季 1 教师 CPD 计划与整个学校目标和评估相关 EC 助教的每学期 CPD 计划包括法定和整个学校目标重点会议 EC 每学期 助教得到支持以完成 HLTA 资格 EC/AHT 正在进行中 新的 ECT 导师有 CPD EC 秋季和夏季 新的入职领导有其角色的 CPD EC 每学期 ECT 完成入职和 ECT 框架,包括释放和指导 EC/AHT 正在进行 TOT 审查,助教评估和增长计划得到支持和时间表 EC/AHT 十月、二月 进行员工 CPD 调查 EC 春季学期

如何建立整个寿命碳基准
1欧洲委员会。 (com(2020)662最终)。 “欧洲的翻新浪潮 - 绿化我们的建筑物,创造就业机会,改善生活”。 可访问:https://eur-lex.europa.eu/legal-content/en/txt/?uri=celex%3A52020DC0662 2欧盟委员会。 (n.d。)。 ''欧洲绿色协议 - 努力成为第一个气候中立大陆'。 可访问:https:// commiss.europa.eu/strategy-and-policy/priorities-2019-2024/european-green-deal_en 3欧盟委员会。 (n.d。)。 ''欧盟排放交易系统(EU ETS)。 可访问:https://climate.ec.europa.eu/eu-action/eu-eu-- emassions-trading-system-system-eu-ets_en 4欧洲委员会。 (n.d。)。 ''建筑产品法规的评论'。 可访问:https://single-market-economy.ec.europa。 EU/soter/struction/struction-products-crenducty-cpr/eview_en 5指令2024/1275。 指令(EU)2024/1275欧洲议会和2024年4月24日理事会关于建筑物的能源绩效(重铸)。 可访问:https://eur-lex.europa.eu/legal-content/en/txt/html/?uri=oj:l_202401275#d1e38-57-11欧洲委员会。(com(2020)662最终)。“欧洲的翻新浪潮 - 绿化我们的建筑物,创造就业机会,改善生活”。可访问:https://eur-lex.europa.eu/legal-content/en/txt/?uri=celex%3A52020DC0662 2欧盟委员会。(n.d。)。''欧洲绿色协议 - 努力成为第一个气候中立大陆'。可访问:https:// commiss.europa.eu/strategy-and-policy/priorities-2019-2024/european-green-deal_en 3欧盟委员会。(n.d。)。''欧盟排放交易系统(EU ETS)。可访问:https://climate.ec.europa.eu/eu-action/eu-eu-- emassions-trading-system-system-eu-ets_en 4欧洲委员会。(n.d。)。''建筑产品法规的评论'。可访问:https://single-market-economy.ec.europa。EU/soter/struction/struction-products-crenducty-cpr/eview_en 5指令2024/1275。指令(EU)2024/1275欧洲议会和2024年4月24日理事会关于建筑物的能源绩效(重铸)。可访问:https://eur-lex.europa.eu/legal-content/en/txt/html/?uri=oj:l_202401275#d1e38-57-1