XiaoMi-AI文件搜索系统
World File Search System和髓样


溶瘤αHerpesvirus和髓样 - 胶质细胞病毒合作增强了全身抗肿瘤反应
MCMV改善了早期时间点KOS-BAC R4.1(DICP47)的复制(图2a)。然而,超过3个DPT,单一疗法和组合组的KOS-BAC R4.1的复制迅速下降。比较组合组与单一疗法组Q-PCR分析显示,肿瘤内KOS-BAC R4.1基因表达和MCMV表达的下调在3个DPT上的下调(图2b)。细胞因子阵列筛选测定法用于确定组合组与单个药物组中肿瘤内细胞因子的相对水平。在3个DPT上,与其他三组相比,在组合组中略微上调了几种促炎性细胞因子(图2C)。相反,在9 dpt的组合组中,各种促炎性细胞因子上调

2024年梅宁抑制剂的更新。一类针对KMT2A重新培养和NPM1突变的急性髓样白血病
摘要:Menin抑制剂是目前正在临床开发中的新型和有前途的药物,其针对HOX/MEIS1转录程序,这对于在组蛋白赖氨酸N-甲基转移酶2A重新培训(KMT2AR)和NPM1-氧化(NPM1)氧化(NPM1M1M1M1MUT)尖锐leukemias中至关重要。这种新型药物的作用机理是基于MENIN – KMT2A复合物的破坏(由染色质重塑蛋白组成),从而导致表达KMT2A或突变NPM1的AML细胞的分化和凋亡。迄今为止,这种新的药物已在I阶段和II期临床试验中进行了测试,无论是单独的,并与协同药物结合使用,在经过预先治疗的急性白血病患者的缓解率和安全性方面,显示出令人鼓舞的结果。在这篇简短的综述中,我们总结了有关梅宁抑制剂的关键发现,重点介绍了有关急性髓细胞性白血病治疗的作用机理和初步的临床数据,尤其是这种有希望的新药物,尤其是Revumenib和Ziftomenib。
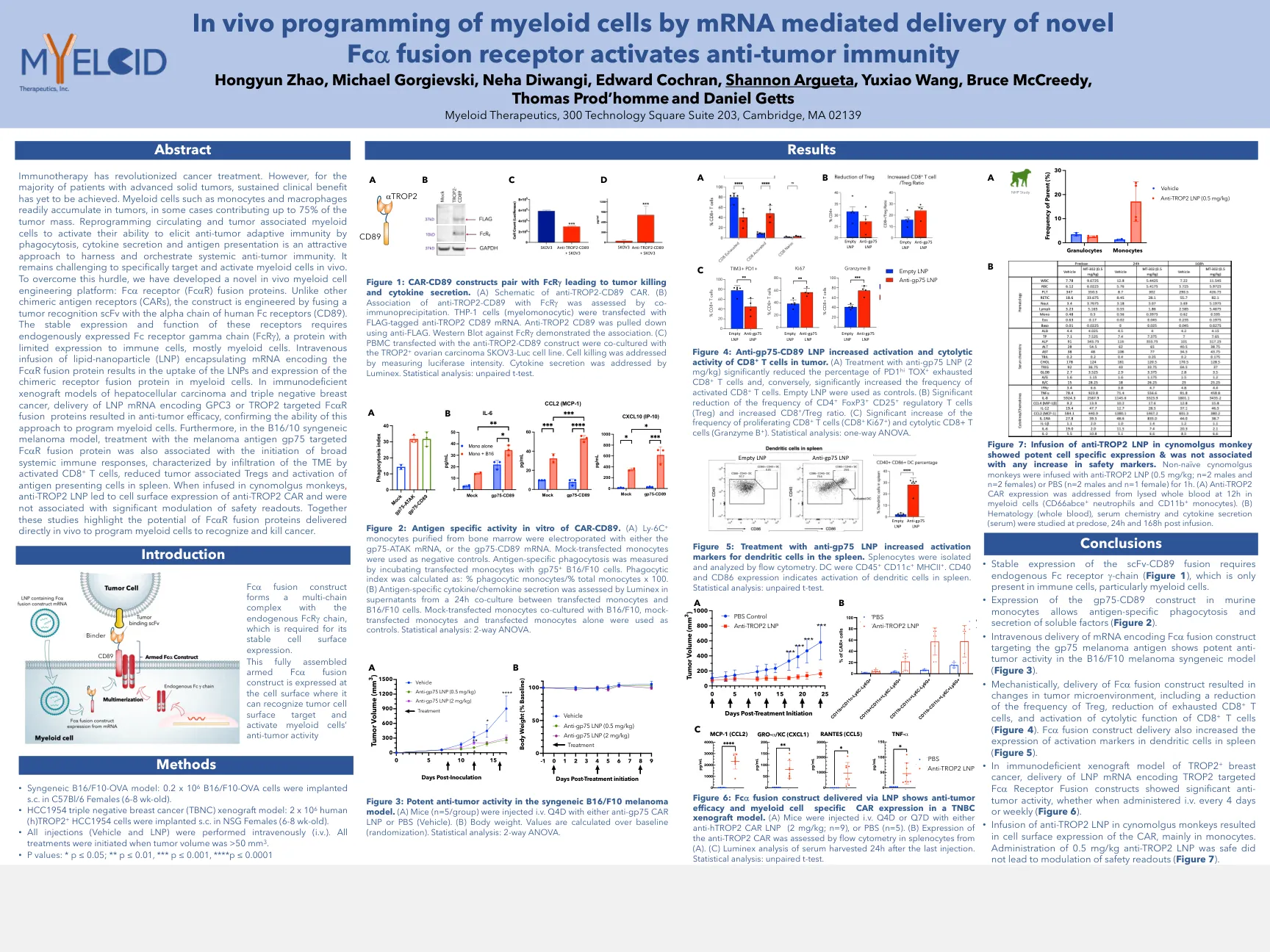
SITC-2022-IN-VIVO-DELIVERY-POSTER.pdf
免疫疗法彻底改变了癌症治疗。但是,对于大多数晚期实体瘤患者,尚未实现持续的临床益处。髓样细胞(如单核细胞和巨噬细胞)很容易积聚在肿瘤中,在某些情况下,肿瘤质量的75%。重编程循环和肿瘤与髓样细胞相关,以激活其通过吞噬作用,细胞因子分泌和抗原表现来激活抗肿瘤适应性免疫的能力,是一种有吸引力的方法,可利用并策划系统性的抗肿瘤免疫。在体内专门靶向和激活髓样细胞仍然具有挑战性。为了克服这一障碍,我们开发了一种新型的体内髓样细胞工程平台:FC A受体(FC A R)融合蛋白。与其他嵌合抗原受体(CAR)不同,该构建体是通过将肿瘤识别SCFV与人体FC受体的α链融合而设计的(CD89)。这些受体的稳定表达和功能需要内源表达的FC受体伽马链(FCR G),这是一种对免疫细胞(主要是髓样细胞)表达有限的蛋白质。术中包裹着编码FC A R融合蛋白的mRNA的脂质纳米颗粒(LNP)导致LNP的摄取并在髓样细胞中摄取嵌合受体融合蛋白的表达。在肝细胞癌的免疫缺陷异种移植模型和三重阴性乳腺癌中,编码GPC3或Trop2靶向FC A R融合蛋白的LNP mRNA的递送导致抗肿瘤疗效,从而确保了这种方法来编程髓样细胞的能力。此外,在B16/10合成性黑色素瘤模型中,用黑色素瘤抗原GP75靶向FC A R融合蛋白的治疗与启动广泛的全身免疫反应的启动,其特征在于激活的CD8 + T细胞通过激活的CD8 + T细胞浸润TME,与肿瘤相关的tregs和Antigen comcipination in Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen of Antigen。当在cynomolgus猴子中注入时,抗Trop2 LNP导致了抗Trop2 Car的细胞表面表达,并且与安全读数的显着调节无关。这些研究共同强调了FC A R融合蛋白直接在体内传递以编程髓样细胞以识别和杀死癌症的潜力。


复发性急性髓样白血病,早期表现为白血病Cutis,对二线治疗难治性
急性髓样白血病(AML)是一种血液学恶性肿瘤,预后不良,通常表现出全身和外牙外表现。白血病Cutis(LC)是白血病细胞进入皮肤的白血病,是AML的罕见且具有临床意义的并发症。 它通常会发出耗尽疾病的参与,可能是在全身复发之前,并且与生存效果不佳有关。 我们报告了一名36岁女性患有AML的案例,她的脸部,躯干和四肢都会在脸部,棕褐色淋巴结损伤中发育,与LC一致。 尽管通过诱导和巩固化疗获得了初步缓解,但她经历了早期的LC复发,全细胞减少症,肝胆功能障碍和感染性并发症,强调了她疾病进展的侵略性。 通过骨髓评估证实了疾病进展,该评估显示60%的爆炸。 尽管进行了强化化疗,抗菌预防和粒细胞群刺激因子的支持,但由于难治性疾病而导致患者的病情恶化。 此案表明LC可能是AML复发的早期标志,强调了及时识别和干预的重要性。 多学科管理和早期诊断重新评估对于改善此类患者的预后至关重要。 需要进一步的研究来探索AML LC患者的新型治疗策略,以增强生存和疾病控制。白血病,是AML的罕见且具有临床意义的并发症。它通常会发出耗尽疾病的参与,可能是在全身复发之前,并且与生存效果不佳有关。我们报告了一名36岁女性患有AML的案例,她的脸部,躯干和四肢都会在脸部,棕褐色淋巴结损伤中发育,与LC一致。尽管通过诱导和巩固化疗获得了初步缓解,但她经历了早期的LC复发,全细胞减少症,肝胆功能障碍和感染性并发症,强调了她疾病进展的侵略性。通过骨髓评估证实了疾病进展,该评估显示60%的爆炸。尽管进行了强化化疗,抗菌预防和粒细胞群刺激因子的支持,但由于难治性疾病而导致患者的病情恶化。此案表明LC可能是AML复发的早期标志,强调了及时识别和干预的重要性。多学科管理和早期诊断重新评估对于改善此类患者的预后至关重要。需要进一步的研究来探索AML LC患者的新型治疗策略,以增强生存和疾病控制。

急性髓样白血病细胞中耐药性FLT3-ITD的双重机制抑制剂控制和药理效用
e661残基。通过细胞热移分析,我们进一步证实了FLT3和KX2-391之间的相互作用。与DMSO相比,熔融曲线有明显的热移。KX2-391治疗导致检测到蛋白质。 KX2-391以剂量依赖性的方式提高了FLT3蛋白的热稳定性。 KX2-391对BA/F3细胞中FLT3具有有效的抑制作用。 它还抑制了表达FLT3ITD的BA/F3的生长以及所有表达FLT3ITD-TKD突变的细胞。 这些细胞以前被称为对AC220等FLT3抑制剂的抗药性。 BA/F3ITD-F691L细胞对KX2- 391(0.032mm vs. 0.372mm)的敏感性提高了十倍。 KX2-391对含有FLT3-ITD(MV4-11,MOLM13)的人类白血病细胞具有更高的抑制作用,比在FLT3-突变的白血病细胞上具有更高的抑制作用。 我们观察到表达FLT3 – ITD,FLT3 – ITD-D835Y和FLT3 – ITD-F691L的BA/F3细胞的剂量依赖性诱导凋亡。 另外,我们在两个FLT3 – ITD阳性AML细胞系中观察到了它(图 1E,F KX2-391显着抑制了FLT3-ITD中的FLT3和下游靶标STAT5,ERK和AKT的磷酸化,FLT3-ITD-F691L-表达BA/F3细胞以及我们的测定面板的其他细胞。 KX2-391是微管蛋白/SRC抑制剂。 我们监测了SRC磷酸化,以评估KX2-391对微管结构的影响。KX2-391治疗导致检测到蛋白质。KX2-391以剂量依赖性的方式提高了FLT3蛋白的热稳定性。KX2-391对BA/F3细胞中FLT3具有有效的抑制作用。 它还抑制了表达FLT3ITD的BA/F3的生长以及所有表达FLT3ITD-TKD突变的细胞。 这些细胞以前被称为对AC220等FLT3抑制剂的抗药性。 BA/F3ITD-F691L细胞对KX2- 391(0.032mm vs. 0.372mm)的敏感性提高了十倍。 KX2-391对含有FLT3-ITD(MV4-11,MOLM13)的人类白血病细胞具有更高的抑制作用,比在FLT3-突变的白血病细胞上具有更高的抑制作用。 我们观察到表达FLT3 – ITD,FLT3 – ITD-D835Y和FLT3 – ITD-F691L的BA/F3细胞的剂量依赖性诱导凋亡。 另外,我们在两个FLT3 – ITD阳性AML细胞系中观察到了它(图 1E,F KX2-391显着抑制了FLT3-ITD中的FLT3和下游靶标STAT5,ERK和AKT的磷酸化,FLT3-ITD-F691L-表达BA/F3细胞以及我们的测定面板的其他细胞。 KX2-391是微管蛋白/SRC抑制剂。 我们监测了SRC磷酸化,以评估KX2-391对微管结构的影响。KX2-391对BA/F3细胞中FLT3具有有效的抑制作用。它还抑制了表达FLT3ITD的BA/F3的生长以及所有表达FLT3ITD-TKD突变的细胞。这些细胞以前被称为对AC220等FLT3抑制剂的抗药性。BA/F3ITD-F691L细胞对KX2- 391(0.032mm vs. 0.372mm)的敏感性提高了十倍。KX2-391对含有FLT3-ITD(MV4-11,MOLM13)的人类白血病细胞具有更高的抑制作用,比在FLT3-突变的白血病细胞上具有更高的抑制作用。我们观察到表达FLT3 – ITD,FLT3 – ITD-D835Y和FLT3 – ITD-F691L的BA/F3细胞的剂量依赖性诱导凋亡。另外,我们在两个FLT3 – ITD阳性AML细胞系中观察到了它(图1E,F KX2-391显着抑制了FLT3-ITD中的FLT3和下游靶标STAT5,ERK和AKT的磷酸化,FLT3-ITD-F691L-表达BA/F3细胞以及我们的测定面板的其他细胞。KX2-391是微管蛋白/SRC抑制剂。 我们监测了SRC磷酸化,以评估KX2-391对微管结构的影响。KX2-391是微管蛋白/SRC抑制剂。我们监测了SRC磷酸化,以评估KX2-391对微管结构的影响。
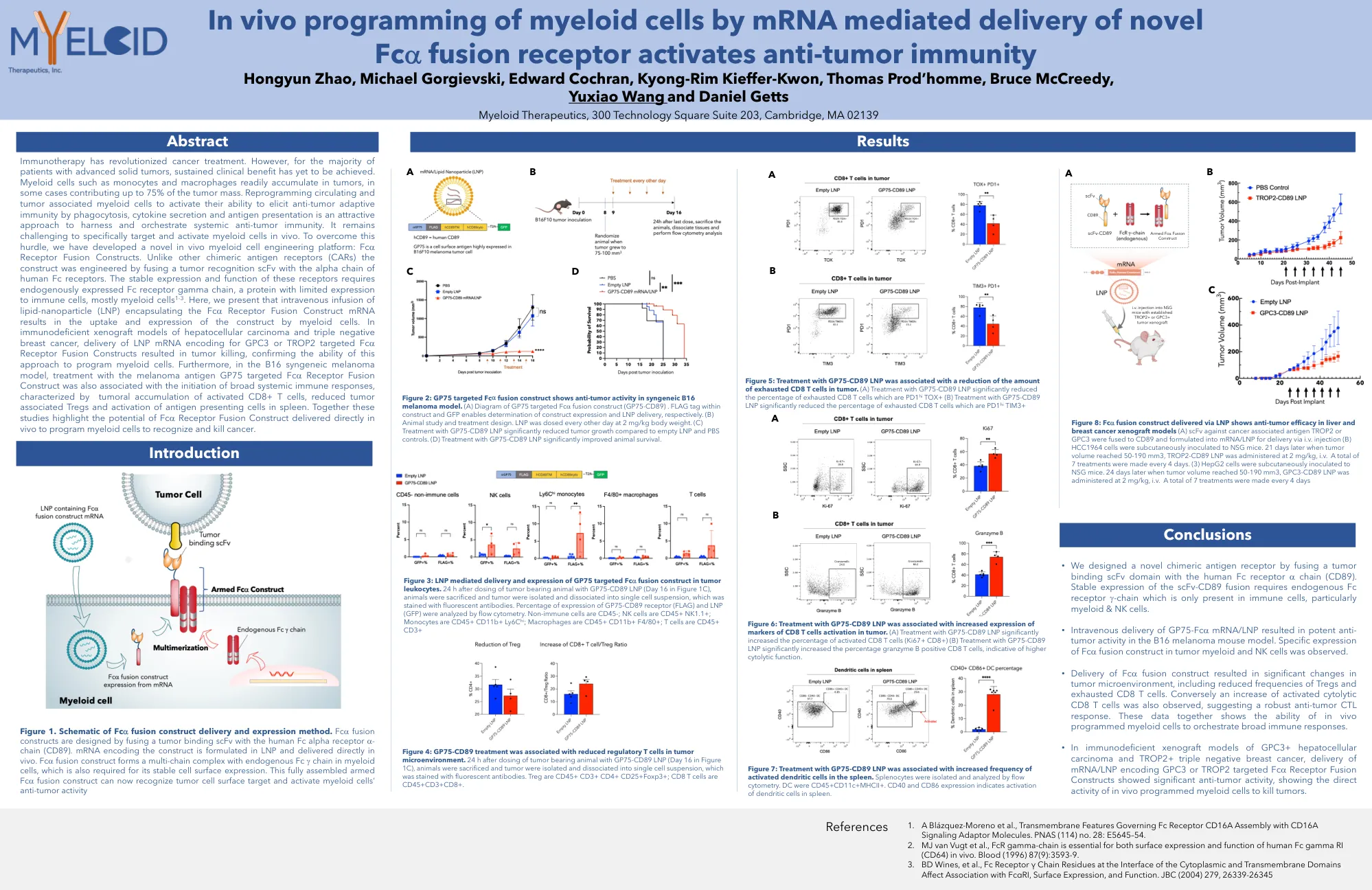
AACR2022体内递送海报
免疫疗法彻底改变了癌症治疗。但是,对于大多数晚期实体瘤患者,尚未实现持续的临床益处。髓样细胞(如单核细胞和巨噬细胞)很容易积聚在肿瘤中,在某些情况下,肿瘤质量的75%。重编程循环和肿瘤与髓样细胞相关,以激活其通过吞噬作用,细胞因子分泌和抗原表现来激活抗肿瘤适应性免疫的能力,是一种有吸引力的方法,可利用并策划系统性的抗肿瘤免疫。在体内专门靶向和激活髓样细胞仍然具有挑战性。为了克服这一障碍,我们开发了一种新型的体内髓细胞工程平台:FC A受体融合构建体。与其他嵌合抗原受体(CAR)不同,该构建体是通过将肿瘤识别SCFV与人体FC受体的α链融合而设计的。这些受体的稳定表达和功能需要内源表达的FC受体γ链,FC受体γ链是一种对免疫细胞表达有限的蛋白质,主要是髓样细胞1-3。在这里,我们介绍了包裹FC A受体融合构建体mRNA的静脉输注脂质 - 纳米颗粒(LNP)导致髓样细胞对构建体的摄取和表达。在肝细胞癌和三重阴性乳腺癌的免疫缺陷异种移植模型中,针对GPC3或trop2靶向FC的LNP mRNA的递送A受体融合构建体导致肿瘤杀死,从而确认了这种方法为骨髓细胞编程的能力。此外,在B16合成性黑色素瘤模型中,用黑色素瘤抗原GP75靶向FC A受体融合构建体的治疗也与启动广泛的全身免疫反应的启动有关,其特征在于肿瘤积累活化的CD8+ T细胞,可减少与肿瘤相关的TREG和SpleeNing spleen and spleen spleen and spleen的活化。这些研究共同强调了FC A受体融合构建体的潜力,直接在体内传递以编程髓样细胞以识别和杀死癌症。

EVI1控制KDM6B介导的组蛋白去甲基化为...
等离子体,单核细胞,中性粒细胞或血小板的增殖增加(1、3、4)。大约30%的被诊断为MD的患者最终患有急性髓样白血病(AML)(5)。eVI1首先被鉴定为具有逆转录病毒诱导的髓样恶质的小鼠中生态病毒整合的常见位点(6)。人类EVI1(MECOM)基因位于Chro-Mosome 3Q26上,EVI1的多种同工型在MECOM基因座(7)中编码。3q26染色体的重排,导致EVI1的上调,经常发生在包括MDS,AML和慢性髓样白血病(CML)在内的髓样恶性疾病中(8-10)。MDS,AML和CML具有INV(3)/T(3; 3)重排通常具有相似的病理特征,预后不良(8、11、12)。It was reported that chromosome rear- rangements cause overexpression of EVI1 due to relocation of enhancers, including GATA binding protein 2 (GATA2) enhancer in inv(3)/t(3;3) (q21q26) (13, 14) and MYC super-enhancer in t(3;8) (q26;q24) close to the EVI1 gene (15).EVI1过表达可能发生在没有3染色体重排的MDS患者中。EVI1上调