XiaoMi-AI文件搜索系统
World File Search System扩增子

从 CRISPR/Cas9 介导的胡萝卜 (Daucus carota subsp. sativus) 着丝粒组蛋白 H3 (CENH3) 基因编辑中获得的见解
单倍体的产生是加速植物育种过程的最有效手段之一。在大多数作物物种中,有效的单倍体技术尚未出现或仅适用于有限的一组基因型。最近发表的关于拟南芥和玉米、小麦等主要谷类作物通过 CRISPR/Cas9 介导的着丝粒组蛋白 H3 基因 (CENH3) 编辑成功诱导单倍体的研究结果表明,这种生产单倍体植物的新方法也可能适用于胡萝卜等蔬菜物种。在这里,我们报告并总结了过去几年专注于基于 CRISPR/Cas9 编辑胡萝卜 CENH3 基因的不同实验和遗传方法。我们还描述了在胡萝卜基因组中发现的第二个 CENH3 基因位点,这使生成和分析假定的单倍体诱导基因型的尝试变得复杂。我们表明,三种不同的 CRISPR/Cas9 靶构建体(单独使用或组合使用)可以成功靶向胡萝卜 CENH3。已经发现了有希望的突变体,例如同框插入/缺失或同框删除突变体,但它们是否能成功用作假定的单倍体诱导物尚不确定。跨越 CRISPR 靶位点的扩增子的下一代测序和基于转录本的扩增子测序似乎是选择有希望的突变体、估计突变频率和首次预测涉及哪个基因的合适方法。本研究的另一个目的是用外来 CENH3 基因同时敲除和补充内源胡萝卜 CENH3 基因。利用根瘤菌将基于 CRISPR/Cas9 的胡萝卜 CENH3 敲除构建体与从人参 (Panax ginseng) 克隆的 CENH3 基因共转化。结果表明,人参 CENH3 蛋白在胡萝卜染色体的着丝粒区域内积累,表明 PgCENH3 可能是这种方法的合适候选者。然而,目前尚不清楚该基因是否在减数分裂细胞分裂过程中充分发挥作用并能够补充致死配子。本文讨论了开发基于 CENH3 的胡萝卜 HI 系统的挑战和未来前景。

NPC中CDC25C的下调干扰了皮质神经发生编辑的原代山羊细胞
遗传改性细胞的基因分型是针对转基因和基因组编辑的至关重要的步骤,例如CRISPR/CAS等系统。检测基因组编辑事件可以与所使用的基因分型方法直接相关,该方法受其成本影响,因为许多实验需要分析大量样品。这项研究的目的是比较基因组DNA(GDNA)提取的直接裂解方法的性能,以检测原代山羊细胞中的敲蛋白和敲除。最初,使用差异量(1,000、5,000和10,000个细胞)和goat Ortiparts(fibroblblasts and fibroblblasts and gote anctermarem Migalsmary Migalmary Migalmary Migalmary Migatiars Migatiars Migatiars)测试了三种GDNA提取方案(方案A,水中的温度A; Prote变性/冻结;小(GAPDH)和大扩增子(HLF转基因)的PCR扩增。所有方案在检测小扩增子方面均成功;但是,在GMEC中,只有协议B仅导致有效的扩增(协议A - 0%,协议B- 93%,协议C- 13.33%,p <0.05)。In a proof- of-principle experiment, the TP53 gene was knocked out in GMECs by CRISPR/Cas9-medi- ated deletion while constructs containing the anti-VEGF monoclonal antibody (pBC-anti- VEGF) and bacterial L-Asparaginase (pBC-ASNase) transgenes were knocked-in sepa- rately in fibroblasts.使用协议B和PCR进行了成功编辑的检测。根据PCR,PBC-ASNase和PBC-Anti-VEGF转基因的整合速率分别为93.6%和72%。使用CRISPR/CAS9对TP53缺失在GMEC中的双重编辑效率为5.4%。我们的结果表明,方案B(热变性/蛋白酶K)可以用作一种廉价且快速的方法,用于检测不同类型的原代山羊细胞中的遗传修饰,其效率率与先前使用提取试剂盒或更复杂的蛋白酶K配方中先前描述的值一致。

CRISPR-CAS裂解测定
研究人员经常依靠Silico CRISPR计算设计算法来产生高性能的GRNA,但仍会在体内经历编辑故障。我们的另一个客户就是这种情况,一个研究团队开发了转基因无菌男性蚊子来打击疟疾的传播。他们的基因编辑实验经常失败,导致大量延迟,每个失败的实验都将项目恢复了8到12个月。在此问题上与他们合作,我们使用CRISPR Analytics平台来量化两个GRNA候选者的扩增子裂解活性。数据表明,两个GRNA都表现出裂解活性在远高于阴性对照的水平上,其中一个GRNA显示出大约是另一个活性的两倍(图2A)。

高效与CRISPR相关的蛋白9核糖核蛋白基于Euglena Gracilis
euglena gracilis是一种单细胞的光养生者,是一种有前途的食物,饲料和生物燃料的材料。但是,该物种中有针对性的诱变方法的发展一直是长期的挑战。在当前的遗传操纵技术中,通过RNP的直接递送进行基因组编辑具有各种优势,包括时间效率,低细胞毒性,高效率和降低距离效应(Jeon等,2017)。在我们的方法,插入和/或缺失(INDEL)突变率为77.7%–90.1%的突变率中,通过在Eggsl2基因中的两个不同靶序列中进行了扩增子测序(Nomura等,2019)。因此,我们在大肠杆菌中开发的基于RNP的基因组编辑开辟了新的途径以揭示基因的功能。

agnėAlminaitė,aistėSerapinaitė,MonikaJazdauskaitė,NeringaDūdaitė,Alexander Klimentov,Dangirašikšnienė,RasaSukackaitėThermo thermo thermo Fisher Sciention
lyo就绪的RPA套件可以成功执行多重RPA反应并在同一反应中扩增几个不同的扩增子。从各种DNA模板(来自1 ng金黄色葡萄球菌基因组DNA中的376 bp)放大了三个靶标,从1 ng的人类基因组DNA中获得了305 bp,从1 ng的人类基因组DNA和238 bp均来自1 ng铜绿假单胞菌基因组DNA的1 ng),四个靶标从各种RNA模板(从各种rna模板中得到1000 000 becies from viruts),从1 ng Chikungunya病毒RNA的含量为253 bp,来自1000份的寨卡病毒RNA和1000份SARS-COV-2 RNA的192 bp)。所有目标均以高特异性检测到没有任何非特异性产品(图5)。

Engen突变检测试剂盒E3321手册
ENGEN突变检测试剂盒提供了用于检测目标基因组编辑事件的试剂。在第一步中,使用Q5热启动High-Fidelity 2X Master Mix放大了来自基因组的靶向区域(即CRISPR/CAS9,TALES,锌指核酸酶)。在变性和重新进行重新进行后,当插入和缺失(Indels)中存在于扩增子池中时,就会形成异质化合物。在第二步中,将退火的PCR产物用Engen T7核酸内切酶I消化,这是一种特定于结构的酶,将识别大于1碱基的不匹配。存在不匹配时切割DNA的两个链,从而导致形成较小的片段。对所得片段的分析提供了基因组编辑实验效率的估计。


解释性信息:对P190或P210定量测定的诊断定性BCR-ABL1分析
方法论:RNA与全血或骨髓分离并反转录。所得的cDNA经过多重PCR扩增,旨在扩增P190,P210或P230 BCR-ABL1融合转录本,涉及ABL1外显子2。ABL1参考基因也被放大以进行标本质量控制并确保RNA的完整性。PCR产物通过毛细管电泳解决,并评估存在表明阳性结果的扩增子的存在。阳性普通P210或P190结果将触发定量P210或P190测试,以提供定量水平作为监测治疗反应的诊断基线。p210的成绩单水平报告为国际量表百分比(%is)。P190转录水平报告为归一化拷贝数(NCN)。这些定量结果被整合到最终报告中。如果初始定性测试为阴性,或者检测到罕见的P230,则不会进行反射测试。
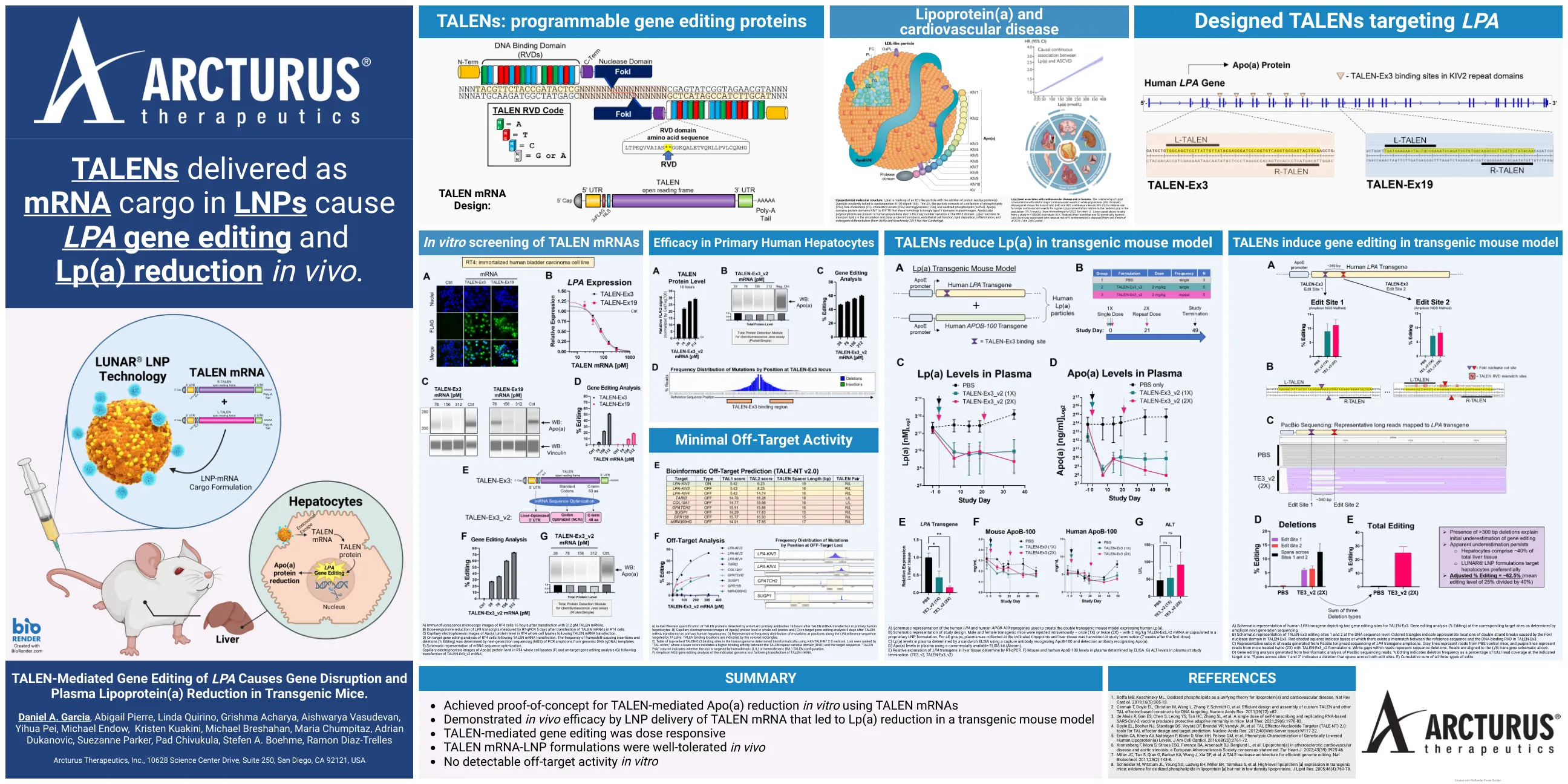
LPA 海报 ASGCT 2023
A) RT4 细胞转染 312 pM TALEN mRNA 16 小时后的免疫荧光显微镜图像。B) RT4 细胞转染 TALEN mRNA 5 天后,通过 RT-qPCR 测量 LPA 转录本的剂量反应性减少。C) TALEN mRNA 转染后 RT4 全细胞裂解物中 Apo(a) 蛋白水平的毛细管电泳图像。D) TALEN mRNA 转染后 RT4 细胞的靶向基因编辑分析。通过基因组 DNA (gDNA) 模板的 PCR 扩增子的下一代测序 (NGS) 确定导致移码的插入和缺失的频率 (编辑百分比)。E) mRNA 序列优化的示意图。转染 TALEN-Ex3_v2 mRNA 后 RT4 全细胞裂解物中 Apo(a) 蛋白水平的毛细管电泳图像 (F) 和靶向基因编辑分析 (G)。

程序性敲除同种异体移植后,光滑双脐螺胚胎细胞系对曼氏血吸虫孢囊的粘附性降低
转染的 Bge 细胞以评估 Cas9 的表达(图 1b)。使用两对引物进行 PCR,以对照或 pCas-BgAIFx4 转染的 Bge 细胞的 cDNA 作为模板,一对引物针对 Cas9,另一对针对 BgActin,后者是 B. glabrata 的肌动蛋白基因,用作参考基因(图 1b、c)。转染后 24 小时检测到瞬时 pCas-BgAIFx4 转染的 Bge 细胞中编码 Cas9 的转录本,并在测定的 9 天内保持表达。在 pCas-BgAIFx4 转染的细胞中观察到 Cas9 mRNA(277 bp)的特异性扩增子,但在未转染的细胞中没有观察到(图 1c)。我们的研究结果支持了先前的研究结果,即揭示了 Bge 细胞中由荧光素酶驱动的 CMV 启动子 [60]。对照参考 BgActin 的表达在 214