XiaoMi-AI文件搜索系统
World File Search System信息学


信息学和计算机技术
Trevor Baylis 是一位发明家。1991 年,他听说了向非洲农村人民提供健康信息的问题。收音机是最好的方式,但人们没有电,买不起昂贵的电池。因此,他发明了一种不需要主电源或电池的收音机。相反,它由弹簧、齿轮和小型发电机组成。那么他的发条收音机实际上是如何工作的呢?当你转动收音机侧面的手柄时,你会卷起弹簧。它与汽车安全带中使用的钢弹簧相同。需要 60 圈才能将弹簧完全卷紧。当弹簧开始松开时,齿轮啮合。有三个 1:10 的增速齿轮。最后一个增速环节是滑轮。滑轮比齿轮运转更安静,因此可以降低噪音。每次第一个齿轮转动时,发电机都会转动一千次。当它转动时,它会发电 - 电压为 3V,电流约为 30 mA。弹簧有足够的能量让收音机运行 30 分钟,然后你才需要再次上发条。全世界有超过两百万台发条收音机在使用。Trevor Baylis 还发明了一种“电动鞋”。它可以在你走路时给电池充电。

计算机和信息学
技术进步导致各种农业系统发生巨大变化,从而大幅提高生产能力 [1]。这些技术进步还确保了粮食安全、肉类和牛奶供应以及工业发展原材料的使用 [1]。农业技术进步越来越多地取代传统农业机械和其他设备的人力和干预 [2]。技术进步促进了农业支持功能的成功自动化,例如机械和肥料的输送以及原材料的生产 [3]。随着计算机技术和计算机系统的发展(表 1),成本进一步降低,农业系统的效率越来越强大 [4, 5]。计算机在农业中的应用分为三个重要领域:图像分析、作物模型和信息技术 [6]。计算机及其在这三个领域的应用改变了大多数传统农业耕作活动的面貌,从农学中最基本的土地利用转变为最高水平的工业加工 [6]。然而,计算机信息系统 (CIS) 是传递农业和非农业部门全球发展所需信息的基本基础 [7, 8]。 CIS 可以看作是人类发展不同领域所需的信息集 [4]。CIS 的主要类型包括执行支持系统 (ESS)、决策支持系统 (DSS)、管理信息系统 (MIS) 和交易处理系统 (TPS) [9]。ESS 通常被称为专家信息系统 (EIS),它结合了 MIS 和 DSS 的诸多功能,信息以根据使用系统的高管的偏好量身定制的形式呈现,例如使用图形用户界面 (GUI) [4]。DSS 向负责对特定情况做出判断的高层管理人员提供信息,并在结构不太完善的情况下(例如风险分析)为决策者提供支持 [4]。

信息学学士学位...
MOOC专业选修课程:这些是各自研究委员会建议的在线课程。仅在VI学期内为学生提供这些课程的详细信息。学生必须仅选择各自BOS建议的课程。在线课程的持续时间应至少12周。学生可以通过主管当局签发的合格证书来完成指定的在线课程。在线课程可以从VI学期开始的任何时间完成,并且仅在VIII学期内考虑学分。课程仅在Swayam - NPTEL平台上提供。本课程获得的学分将不考虑荣誉学位计划下的学分。

健康信息学
第 150 卷。K.-P. Adlassnig、B. Blobel、J. Mantas 和 I. Masic(编辑),联合健康欧洲的医学信息学 - MIE 2009 论文集 - 欧洲医学信息学联合会第 22 届国际大会第 149 卷。RG Bushko(编辑),健康未来战略第 148 卷。R. Beuscart、W. Hackl 和 C. Nøhr(编辑),药物不良事件的检测和预防 - 信息技术和人为因素第 147 卷。T. Solomonides、M. Hofmann-Apitius、M. Freudigmann、SC Semler、Y. Legré 和 M. Kratz(编辑),健康网格研究、创新和商业案例 - HealthGrid 2009 论文集第 146 卷。K. Saranto 等人(Eds.),连接健康与人类 - NI2009 论文集 - 第 10 届国际护理信息学大会第 145 卷。A. Gaggioli 等人 (Eds.),康复领域的先进技术 - 通过虚拟现实、机器人、可穿戴系统和脑机接口增强认知、身体、社交和沟通能力第 144 卷。BK Wiederhold 和 G. Riva (Eds.),网络治疗和远程医疗年度回顾 2009 - 行为社会和神经科学领域的先进技术第 143 卷。JG McDaniel (Ed.),健康领域信息技术和通信的发展



1信息学简介
信息学是一个非常年轻的科学学科和学术领域。对术语的解释(从现代欧洲科学文献中使用)尚未建立并普遍接受[1]。大多数现代计算机和计算机技术的祖国位于美利坚合众国,这就是为什么美国信息学术语与欧洲交织在一起的原因。美国术语计算机科学被认为是信息学术语的同义词,但是这两个术语具有不同的历史,含义有所不同,并且是构思树的根源,这些概念树充满了不同的术语。虽然计算机科学领域的专家被称为计算机工程师,但信息学的从业者可能被称为信息医生。计算机科学一词的历史始于1959年,当时路易斯·菲恩(Louis Fein)主张创建与哈佛商学院相似的计算机科学研究生院。在证明学校的名称时,他提到了管理科学,就像计算机科学具有应用和跨学科的性质,并且具有学科的特征。尽管名称(计算机科学),但与计算机相关的大多数科学领域都不包括计算机本身的研究。结果,在英语世界中提出了几种替代名称,例如,一些主要大学的一些学院更喜欢计算科学一词,而是强调术语之间的差异。卡尔·斯坦布赫(Karl Stebuch)于1957年推出了德国术语Informatik,菲利普·德雷福斯(Philippe Dreyfus)于1962年推出了法国术语Informatique。彼得·诺尔(Peter Naur)建议了斯堪的纳维亚术语数据法,以反映科学学科运作和处理数据的事实,尽管不一定是使用计算机的使用。英语术语信息学被创造为两个单词的组合:信息和自动化;最初,它描述了信息的自动处理科学。信息学的核心概念是信息的转换,这是通过生物和人工制品的计算和通信进行的。信息转换使其用于决策。
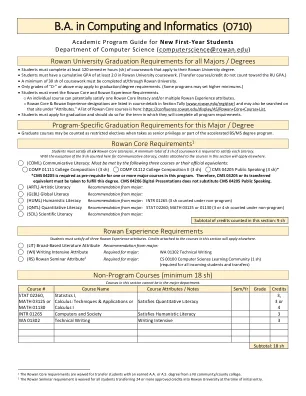
B.A.在计算和信息学(O710)B.A.在计算和信息学(O710)
学生必须满足所有六个罗文核心文学。至少需要总共3 sh的课程才能满足每个识字能力。除了在这里计算的9 sh以外的交流素养之外,本节中有关课程附加的学分将适用于其他地方。⃝(COML)交流素养:必须通过以下三门课程或其官方等效来满足:⃝comp 01111大学作曲I(3 sh)⃝comp 01112大学组成II(3 sh)⃝CMS04205公开演讲(3 sh) * * CMS 04205作为一个或更多主要校长的先决条件。因此,必须采用CMS 04205或其转移的等效物以实现此学位。CMS 04206数字演示文稿不能代替CMS 04205公开演讲。⃝(ARTL)专业的艺术素养建议:⃝(GLBL)专业的全球识字率建议:⃝(HUML)Major:Inter 01265(Inter 01265)的人文素养建议(3 sh,根据非计划)⃝(QNTL)⃝(QNTL)量(QNTL)定量素养建议来自Major:STAT 02260; MATH 03125或01130(根据非程序计数计数3或4 SH)(SCIL)科学素养建议:

量子化学和化学信息学...
一个世纪前,量子力学诞生时,狄拉克声称发现了化学的基本原理,即原子和分子水平上的材料科学——但他也承认,要将其全面应用,需要开发有效的计算技术。接下来的十年记录了信息科学的诞生(冯·诺依曼和维格纳是这两门科学的创始人之一):化学的发展和应用变得至关重要,如今已经成熟:量子化学解释和预测了在行星大气和星际介质的稀薄环境中发生的各种新现象,包括与热和非平衡等离子体相关的现象;新兴任务被强加给生物化学家,这对生命和健康科学来说是必需的工具;固体导体和半导体的电磁特性在光电应用方面的研究十分活跃;当前的圣杯是支持量子计算开发的化学硬件,微观、中观和宏观尺度的物理化学模型可以让人们积累大量数据——它们只能通过化学信息学方法来处理,以审查材料或分子的性质;既利用强大的机器学习方法获取原本无法获得的信息,又通过人工智能方法揭示行为的隐藏相关性和普遍性,而这些在当前复杂性理论的非线性方程中是模糊的。