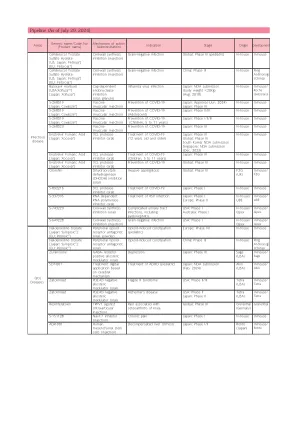
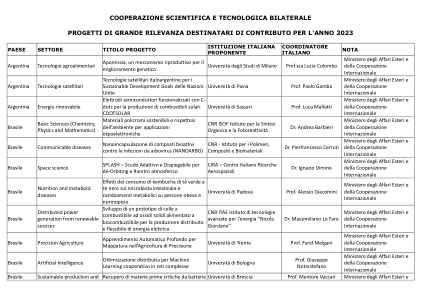
XiaoMi-AI文件搜索系统
World File Search System变构


变构线粒体CLPP激动剂ONC206改变了压力反应,代谢和表观遗传学特征,以在高级GL
参考文献1。Allen Je和Al。SCI Transl Med。2013; 5(171):1717; 2。 rb冻结和al。 Pharmacol's。 2021; 100:372-387; 3。 ns疯狂和al。 nat公社。 2019; 10:5221; 4。 Ishizawa J和Al。 癌细胞。 2019; 35:721-737 E9; 5。 PR仪式和Al。 ACS头。 2019; 14:1020-1029; 6。 chi as和al。 j神经。 2019; 145(1):97-105; 7。 theeler bj和al。 J Clin Oncol.2020; 8。 vv prabhu和al。 歌手res。 2020; 80(16_supplementary):5688-5688; 9。 Wagner J和Al。 循环单元。 2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2013; 5(171):1717; 2。rb冻结和al。Pharmacol's。2021; 100:372-387; 3。ns疯狂和al。nat公社。2019; 10:5221; 4。 Ishizawa J和Al。 癌细胞。 2019; 35:721-737 E9; 5。 PR仪式和Al。 ACS头。 2019; 14:1020-1029; 6。 chi as和al。 j神经。 2019; 145(1):97-105; 7。 theeler bj和al。 J Clin Oncol.2020; 8。 vv prabhu和al。 歌手res。 2020; 80(16_supplementary):5688-5688; 9。 Wagner J和Al。 循环单元。 2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2019; 10:5221; 4。Ishizawa J和Al。癌细胞。2019; 35:721-737 E9; 5。 PR仪式和Al。 ACS头。 2019; 14:1020-1029; 6。 chi as和al。 j神经。 2019; 145(1):97-105; 7。 theeler bj和al。 J Clin Oncol.2020; 8。 vv prabhu和al。 歌手res。 2020; 80(16_supplementary):5688-5688; 9。 Wagner J和Al。 循环单元。 2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2019; 35:721-737 E9; 5。PR仪式和Al。ACS头。2019; 14:1020-1029; 6。 chi as和al。 j神经。 2019; 145(1):97-105; 7。 theeler bj和al。 J Clin Oncol.2020; 8。 vv prabhu和al。 歌手res。 2020; 80(16_supplementary):5688-5688; 9。 Wagner J和Al。 循环单元。 2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2019; 14:1020-1029; 6。chi as和al。j神经。2019; 145(1):97-105; 7。 theeler bj和al。 J Clin Oncol.2020; 8。 vv prabhu和al。 歌手res。 2020; 80(16_supplementary):5688-5688; 9。 Wagner J和Al。 循环单元。 2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2019; 145(1):97-105; 7。theeler bj和al。J Clin Oncol.2020; 8。vv prabhu和al。歌手res。2020; 80(16_supplementary):5688-5688; 9。Wagner J和Al。循环单元。2017; 16:1790-1799; 10。 Staley A和Al。 AM J Singing Res。 2021; 11(11):5374-5387; 11。 张Y和Al。 Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。2017; 16:1790-1799; 10。Staley A和Al。AM J Singing Res。2021; 11(11):5374-5387; 11。张Y和Al。Oncol Front。 2020; 10:57141; 12。 Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。Oncol Front。2020; 10:57141; 12。Tucker K和Al。 AM J Singing Res。 2022; 12(2):521-536; 13。 vv prabhu和al。 Clins Ress。 2019; 25:2305-2313; 14。 Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。Tucker K和Al。AM J Singing Res。2022; 12(2):521-536; 13。vv prabhu和al。Clins Ress。2019; 25:2305-2313; 14。Jassal B和Al。 九十res。 2020 JAN 8; 48(D1):D498-D503。Jassal B和Al。九十res。2020 JAN 8; 48(D1):D498-D503。2020 JAN 8; 48(D1):D498-D503。

核糖核苷酸还原酶,淋病的新药物
抽象抗生素耐药酸酯(NG)是由于增加的多药耐药性(MDR)生物的增加而成为新兴的公共卫生威胁。我们确定了两个新型的口服活性抑制剂PTC-847和PTC-672,它们表现出狭窄的活性范围,包括NG,包括MDR分离株。通过选择对新型抑制剂有抗性的生物并测序其基因组,我们确定了一个新的治疗靶标,即IA核糖核苷酸还原酶(RNR)。在Ng MAP中分解突变与α亚基的N末端锥结构域,我们在这里显示的是在存在β亚基和变构效应子DatP的情况下形成抑制的α4β4状态。 酶测定确认PTC-847和PTC-672抑制NG RNR,并揭示了变构效应器DATP增强了抑制作用。 口服PTC-672的口服降低了小鼠模型中的NG感染,并且可能具有治疗对当前药物具有抗药性的治疗的治疗潜力。分解突变与α亚基的N末端锥结构域,我们在这里显示的是在存在β亚基和变构效应子DatP的情况下形成抑制的α4β4状态。酶测定确认PTC-847和PTC-672抑制NG RNR,并揭示了变构效应器DATP增强了抑制作用。口服PTC-672的口服降低了小鼠模型中的NG感染,并且可能具有治疗对当前药物具有抗药性的治疗的治疗潜力。口服PTC-672的口服降低了小鼠模型中的NG感染,并且可能具有治疗对当前药物具有抗药性的治疗的治疗潜力。

氧化依布硒及其衍生物是用于治疗 HER2 阳性癌症的新型变构 HER2 抑制剂
乳腺癌。然而,相关的耐药机制和毒性凸显了对这些癌症的新治疗方法的需求。我们最近发现,在正常细胞中,HER2 通过与埃兹蛋白/根蛋白/膜突蛋白 (ERM) 家族成员直接相互作用而稳定在催化抑制状态。在 HER2 过表达的肿瘤中,膜突蛋白的低表达导致 HER2 的异常激活。通过旨在寻找膜突蛋白模拟化合物的筛选,我们鉴定出依布硒氧化物。我们表明,依布硒氧化物和一些衍生物可有效抑制过表达的 HER2 以及突变和截短的致癌形式的 HER2,这些形式对目前的疗法具有抗性。依布硒氧化物选择性抑制 HER2 + 癌细胞的锚定依赖性和非锚定依赖性增殖,并与目前的抗 HER2 治疗药物联合使用具有显著的益处。最后,依布硒氧化物显著抑制了体内 HER2 + 乳腺肿瘤的进展。总之,这些数据证明依布硒氧化物是一种新发现的 HER2 变构抑制剂,可用于 HER2 + 癌症的治疗干预。

DNA编码的库筛选发现针对隐性变构结合位点的有效DNMT2抑制剂
。cc-by-nc-nd 4.0国际许可证。根据作者/资助者,它是根据预印本提供的(未经同行评审的认证),他已授予Biorxiv的许可证,以在2025年1月16日发布的此版本中在版权所有者中显示预印本。 https://doi.org/10.1101/2025.01.15.632061 doi:Biorxiv Preprint

KSQ-4279:一种用于治疗具有同源重组缺陷癌症的第一类USP1抑制剂
•KSQ-4279是一种可逆的,具有ki = 1.2nm的USP1的变构抑制剂•ksq-4279绑定和未结合的USP1结构均已解决,揭示了诱导的拟合机构

引用纸浆版本(APA):Cleary,S。R.,Seflova,J.,Cho,E.E.,E.E.,Bisht,K.,Khandelia,H.,Espinoza-Fonseca,L.M。,L. M.,&Robia,S.L。(2024)。
磷兰班(PLB)是一种跨膜小肽,可调节心脏肌肉中的肌质网Ca 2+ -ATPase(SERCA),但这种调节的物理机制仍然很熟悉。PLB降低了活性SERCA的Ca 2+敏感性,从而增加了泵循环所需的Ca 2+浓度。然而,当不存在ATP时,PLB不会降低Ca 2+与SERCA的结合,这表明PLB不会抑制SERCA Ca 2+ afintient。对这些看似冲突的结果的主要解释是,PLB在与Ca 2+结合相关的SERCA酶促循环中的转变减慢了转运Ca 2+的依赖性,而不会实际影响Ca 2+协调位点的等电数。在这里,我们考虑了另一个假设,即在没有ATP的情况下,Ca 2+结合的测量可忽略核苷酸结合的重要变构效应,从而增加了SERCA Ca 2+结合效果。我们推测PLB通过逆转这种同义来抑制SERCA。为了测试这一点,我们使用了荧光的SERCA生物传感器来量化非循环SERCA的Ca 2+在存在和不存在不可用的ATP-ANALOG AMPPCP的情况下。核苷酸激活增加了SERCA Ca 2+的原性,并且通过PLB的共表达逆转了这种效果。有趣的是,在没有核苷酸的情况下,PLB对Ca 2+的原性没有影响。这些结果调解了先前的ATPase分析与Ca 2+结合测定的冲突观察结果。此外,SERCA的结构分析揭示了连接ATP和Ca 2+结合位点的新型变构途径。我们提出的这一途径被PLB结合所破坏。因此,PLB通过通过ATP中断泵的变构激活而降低了SERCA的平衡Ca 2+。因此,PLB通过通过ATP中断泵的变构激活而降低了SERCA的平衡Ca 2+。

选择性变构TYK2小分子抑制剂可调节免疫细胞功能,并改善实验性自身免疫性脑脊髓炎。
我们确定了生化测定中A-005结合对TYK2调节(JH2)和激酶(JH1)域的亲和力。在商业激酶面板中评估了化合物的效力和选择性。在人外周血单核细胞(PBMC),全血和小胶质细胞中评估了该化合物对免疫细胞活性的影响。脑脊液(CSF)暴露,并在大鼠中进行微透析,以评估化合物越过血脑屏障的潜力。最后,我们评估了该化合物对小鼠EAE临床体征的影响。› A-005是一种高度有效的变构小分子TYK2抑制剂,预计将在2024年初进行人类临床试验。› A-005抑制人类全血,PBMC和小胶质细胞的TYK2途径激活。›大鼠的一项微透析研究显示了A-005越过血脑屏障的能力。› A-005在预防或治疗时降低EAE临床评分。› PH1临床试验中的剂量水平预计将在中枢神经系统和外围实现完全靶标的抑制作用。