XiaoMi-AI文件搜索系统
World File Search System基因座
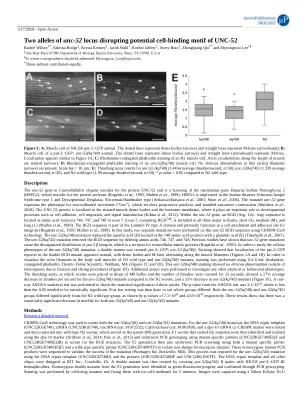
unc-52 基因座的两个等位基因破坏了潜在的细胞......
图 1:A:NK358 pat-3::GFP 动物的肌肉细胞。虚线代表致密体(箭头),直线代表 M 线(箭头);B:pat-3::GFP; unc-52(kq748) 动物的肌肉细胞。虚线代表致密体(箭头),直线(箭头)代表 M 线。定位看起来与图 1A 相似;C:N2 肌肉细胞的罗丹明偶联鬼笔环肽染色。沿肌肉长度的肌动蛋白细胞骨架被染色(箭头);D:unc-52(kq748) 肌肉细胞的罗丹明偶联鬼笔环肽染色。细(肌动蛋白)丝(箭头)中没有明显异常。比例尺 = 10 µm。; E:unc-52 (kq748)(平均每秒 1.4454 次冲击,n=50)、unc-52(kq745)(平均每秒 1.339 次冲击,n=50)和 N2 野生型(平均每秒 1.99 次冲击,n=50)的冲击试验结果。 * 与 N2 野生型相比,p 值 < 0.05。

小鼠基因组重写和三个重要疾病基因座的定制
基因工程小鼠模型 (GEMM) 有助于我们了解人类病理并开发新疗法,但在小鼠身上忠实地重现人类疾病却具有挑战性。基因组学的进展凸显了非编码调控基因组序列的重要性,这些序列控制着许多人类疾病的时空基因表达模式和剪接 1,2 。包括需要大规模基因组工程的调控大范围基因组区域应该可以提高疾病建模的质量。现有方法限制了 DNA 传递的大小和效率,阻碍了我们称之为基因组重写和定制 GEMM(GREAT-GEMM)的高度信息模型的常规创建。在这里,我们描述了 8 哺乳动物逐步切换抗生素抗性标记以进行整合 9 (mSwAP-In),这是一种在小鼠胚胎干细胞中进行高效基因组重写的方法。我们展示了使用 mSwAP-In 对定制的 Trp53 基因座进行多达 115)kb 的迭代基因组重写,以及使用 116)kb 和 180)kb 人类 ACE2 基因座对小鼠进行人源化。ACE2 模型重现了人类 ACE2 的表达模式和剪接,值得注意的是,与现有的 K18-hACE2 模型相比,在受到 SARS-CoV-2 攻击时表现出的症状较轻,因此代表了一种更像人类的感染模型。最后,我们通过在 ACE2 GREAT-GEMM 中对小鼠 Tmprss2 进行双等位基因人源化,展示了连续基因组写入,突出了 mSwAP-In 在基因组写入方面的多功能性。

linkAgeMapView:图链接组映射与定量性状基因座
lmv.linkage.plot(mapthis,outfile,mapthese = null,at.axis = null,autoconnadj = true,cex.axis = par(“ cex.axis”),cex.lgtitle = par = par(“ cex.main” col.lgtitle = par(“ col.main”),col.main = par(“ col.main”),conndf = null,denmap = false,dupnbr = false,font.axis = par(“ font.axis”),font.lgtitle = par(“ 0.3,labels.axis = true,lcex = par(“ cex”),lcol = par(“ col”),lfont = par(“ font”),lgperrow = null,lgtitles = null,lgw = 0.25,lgw = 0.25,lg.col = null = null,lg.lwd = par( lwd.ticks.axis = lwd.axis, main = NULL, markerformatlist = NULL, maxnbrcolsfordups = 3, pdf.bg = "transparent", pdf.family = "Helvetica", pdf.fg = "black", pdf.width = NULL, pdf.height = NULL, pdf.pointsize = 12, pdf.title = "LinkageMapView R output", posonleft = NULL, prtlgtitles = TRUE, qtldf = NULL, revthese = NULL, rcex = par("cex"), rcol = par("col"), rfont = par("font"), roundpos = 1, rsegcol = TRUE, ruler = FALSE, sectcoldf = NULL, segcol = null,qtlscanone = null,showonly = null,unt =“ cm”,ylab = units)

在半sib中的定量性状基因座的多个标记映射在半sib中的定量性状基因座的多个标记映射
许多作者考虑了用于分析来自杂种种群数据的设计(例如Neimann-Sprensen和Robertson,1961年; Soller和Genizi,1978年; Geldermann等,1985; Weller等,1990)。这些方法的缺点是他们一次使用来自单个MARIRW的信息。没有标记将具有统一性的杂合性,因此对于任何给定的标记,有些父亲都会是纯合的,因此是非信息的。这会浪费信息,并在QTL的估计位置中引入偏差可能会有更大的问题。此外,提出的最小二乘方法不能单独估计任何检测到的QTL的位置和效果。最大似然(ML)方法(Weller,1986; Knott and Haley,1992a)可以估计这两种效果,但是通常仅使用单个标记(Weller,1986; Knott; Knott and Haley,1992a and B)估计,位置与标记相对(I.E.可以是它的任何一侧)。

定量性状基因座,G×E和G×G的血糖特质
竞争利益这项研究的赞助商在指导委员会中代表,并在研究设计,研究方式和出版中发挥了作用。尽管指导委员会的所有成员都对报告的内容投入了,但资助机构并未在写作小组中代表。写作组中的所有作者都可以访问所有数据。表达的意见是调查人员的意见,不一定反映了资金机构的观点。资助者在研究设计,数据收集,分析,发布或准备手稿中没有作用。在出版时,KJM是Eli Lilly and Company的雇员。数据收集是在此工作之前发生的,数据分析和手稿准备工作独立于Eli Lilly and Company进行。其他作者声明没有利益冲突。

在小麦(Triticum aestivum l.)中,在染色体2D上,稳定的主要效应定量性状基因座的稳定效应定量特征基因座的识别和验证和验证
摘要:一个重组的近交系数量,包括371条线,由每个尖峰(KNP)基因型T1208和低KNPS基因型Chuannong18(CN18)开发。由小麦55k SNP阵列构建的遗传连锁图由11,583个标记组成。在三年内检测到与KNP有关的定量性状基因座(QTL)。分别使用ICIM-BIP,ICIM-MET和ICIM-EPI方法来识别八个,二十七个和四个QTL。一个QKTL,QKNPS.SAU-2D.1,在染色体2D上映射,可以平均解释18.10%的表型变化(PVE),并被视为KNP的主要稳定QTL。此QTL位于2D染色体上的0.89 MB间隔,并由标记物AX-109283238和AX-111606890倾斜。此外,设计了与qknps.sau-2d.1紧密相关的Kompetive Primentififififif PCR(KASP)标记的KASP-AX-111462389。QKNPS.SAU-2D.1对KNP的遗传作用成功地确认了两个RIL种群。结果还表明,KNPS和1000个内核重量(TKW)的显着增加是由QKNPS.SAU-2D.1引起的,这是由于尖峰数量(SN)的减少而克服了劣势,并最终导致晶粒产量的显着增加。此外,在QKNPS.SAU-2D.1位于中国春季参考基因组中的间隔内,仅发现了十五个基因,并且两个可能与KNP相关的基因都被鉴定出来。qknps.sau-2d.1可能会为未来的高产小麦育种提供新的资源。

乳突的多符合元分析发现了各种不同的敏感性基因座
保留所有权利。未经许可就不允许重复使用。(未经同行评审证明)是作者/资助者,他已授予Medrxiv的许可证,以永久显示预印本。此预印本版的版权持有人于2025年1月31日发布。 https://doi.org/10.1101/2025.01.28.25321288 doi:medrxiv preprint

CRISPR/Cas9 介导大转基因整合至猪 CEP112 基因座
摘要 成簇的规律间隔短回文重复序列 (CRISPR) 相关蛋白 9 (Cas9) 是一种精确的基因组操作工具,可在各种细胞和生物体中产生靶向基因突变。尽管 CRISPR/Cas9 可以有效地产生基因敲除,但同源定向修复介导的基因敲入 (KI) 效率仍然很低,尤其是对于大片段整合。在本研究中,我们建立了一种有效的方法,用于 CRISPR/Cas9 介导的大型转基因盒整合,该盒携带唾液腺表达的多种消化酶 (20 kbp) 在猪胎儿成纤维细胞 (PFF) 的 CEP112 基因座中。我们的结果表明,使用具有短臂和长臂的最佳同源供体在 CEP112 基因座中产生了最好的 CRISPR/Cas9 介导的 KI 效率,并且 CEP112 基因座的靶向效率高于 ROSA26 基因座。CEP112 KI 细胞系被用作核供体,进行体细胞核移植以创建转基因猪。我们发现 KI 猪 (705) 在唾液腺中成功表达三种微生物酶 (b-葡聚糖酶、木聚糖酶和植酸酶)。这一发现表明 CEP112 基因座通过组织特异性启动子支持外源基因表达。总之,我们利用我们的最佳同源臂系统成功地在猪体内靶向 CEP112 基因座,并建立了一种改良的猪唾液中表达外来消化酶的模型。

反复进化和选择形成了淀粉酶基因座的结构多样性
农业的采用引发了人类饮食向富含淀粉的快速转变 1 。淀粉酶基因有助于淀粉的消化,在一些高淀粉摄入量的现代人类群体中观察到了淀粉酶拷贝数的增加 2 ,尽管缺乏近期选择的证据 3,4 。在这里,使用来自大约 5,600 个当代和古代人类的 94 个长读单倍型解析组装和短读数据,我们解决了淀粉酶基因座结构变异的多样性和进化历史。我们发现淀粉酶基因在农业群体中的拷贝数高于渔猎和游牧群体。我们鉴定了 28 种不同的淀粉酶结构架构,并证明在整个人类近代历史中,几乎相同的结构在不同的单倍型背景下反复出现。 AMY1 和 AMY2A 基因均经历了多次重复/缺失事件,突变率高达单核苷酸多态性突变率的 10,000 倍以上,而 AMY2B 基因重复则具有单一起源。使用基于泛基因组的方法,我们推断了数千名人类的结构单倍型,并在现代农业人群中以更高的频率识别出大量重复的单倍型。利用 533 个古人类基因组,我们发现,在过去 12,000 年中,西欧亚大陆中含有重复的单倍型(基因拷贝数多于祖先单倍型)的频率迅速增加,这表明存在正向选择。总之,我们的研究强调了农业革命对人类基因组的潜在影响以及结构变异在人类适应中的重要性。

定量性状基因座的分子映射,以抵抗西红柿早期疫病
早期疫病(EB),由linariae(Neerg。)(SYN。A。tomatophila)Simmons是一种影响世界各地的西红柿(Solanum lycopersicum L.)的疾病,具有巨大的经济影响。本研究的目的是绘制与西红柿中EB耐药性相关的定量性状基因座(QTL)。F 2和F 2:3的映射种群由174条线组成,这些群体在2011年的自然条件下评估了NC 1celbr(抗性)×Fla。7775(易感性),并通过人工接种在2015年的温室中进行了自然条件评估。总共使用了375个具有特定PCR(KASP)测定法的基因分型父母和F 2种群的分析。表型数据的广泛遗传力估计为2011年和2015年的疾病评估分别为28.3%和25.3%。QTL分析显示,六个QTL与染色体2、8和11(LOD 4.0至9.1)上的EB抗性相关,解释了3.8至21.0%的表型变异。这些结果表明,NC 1celbr中EB耐药性的遗传控制是多基因的。这项研究可能有助于将EB抗性QTL和标记辅助选择(MAS)进一步绘制,以将EB耐药基因转移到精英番茄品种中,包括扩大番茄中EB耐药性的遗传多样性。