XiaoMi-AI文件搜索系统
World File Search System生物制品
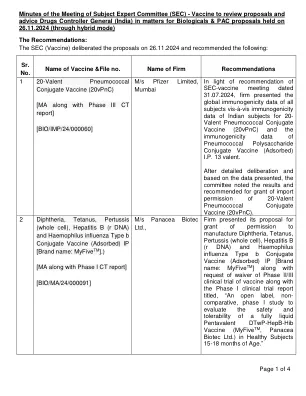
疫苗主题专家委员会 (SEC) 会议纪要,审查印度药品管理局关于生物制品问题的提案和建议
根据 2024 年 7 月 31 日 SEC 疫苗会议的建议,公司提交了所有受试者的全球免疫原性数据,以及印度受试者的 20 价肺炎球菌结合疫苗 (20vPnC) 的免疫原性数据和肺炎球菌多糖结合疫苗 (吸附) IP 13 价的免疫原性数据。经过详细审议并根据提交的数据,委员会注意到了结果并建议授予 20 价肺炎球菌结合疫苗 (20vPnC) 的进口许可。 2 白喉、破伤风、百日咳(全细胞)、乙型肝炎(r DNA)和b型流感嗜血杆菌结合疫苗(吸附)IP [品牌名称:MyFive TM 。)[MA 以及 I 期 CT 报告] [BIO/MA/24/000091]

抗菌和抗真菌药物的有限群体途径
1 本指南由美国食品药品管理局药品评价与研究中心(CDER)和生物制品评价与研究中心(CBER)传染病办公室和新药政策办公室制定。2 公法 114-255、130 Stat. 1033 (2016) (21 USC 356)。3 在本指南中,除非另有规定,所有对药品、药品和产品的引用均包括 CDER 和 CBER 监管的人用药品和生物制品。4 在本指南中,申办方和申请人一词包括任何根据《联邦食品、药品和化妆品法》第 505 节或《公共卫生服务法》第 351 节提出的新药临床试验申请 (IND) 的申办方或新药申请或生物制品许可申请的申请人。


疫苗和相关生物制品咨询委员会 2023 年 5 月 18 日会议简报文件 - FDA
附件包含美国食品药品管理局 (FDA) 为疫苗和相关生物制品咨询委员会小组成员准备的背景信息。FDA 背景资料包通常包含由 FDA 个别审阅者撰写的评估和/或结论和建议。此类结论和建议不一定代表个别审阅者的最终立场,也不一定代表审查部门或办公室的最终立场。我们已向美国食品药品管理局 (FDA) 提交了一份生物制品许可申请 (BLA),以支持 RSVpreF (Abrysvo) 的许可,其拟议的适应症和用途是“通过对孕妇进行主动免疫,预防从出生到 6 个月大的婴儿呼吸道合胞病毒 (RSV) 引起的下呼吸道疾病 (LRTD) 和严重 LRTD”,提交给本咨询委员会,以获得委员会的见解和意见,背景资料包可能不包括与最终监管建议相关的所有问题,而是旨在重点关注机构确定的供咨询委员会讨论的问题。在考虑了咨询委员会的意见并完成了所有审查之前,FDA 不会就手头的问题做出最终决定。最终决定可能会受到咨询委员会会议上未讨论的问题的影响。

疫苗和相关生物制品咨询委员会 2021 年 6 月 10 日会议简报文件 - FDA
词汇表 BEST 生物制品有效性和安全性 BLA 生物制品许可申请 CBRN 化学、生物、放射或核 CFR 联邦法规 CMC 化学、制造和控制 CMS 医疗保险和医疗补助服务中心 COVID-19 2019 年冠状病毒病 EUA 紧急使用授权 FDA 食品药品管理局 GMT 几何平均滴度 HHS 美国卫生与公众服务部 IND 研究性新药 PMC 许可后承诺 PMR 许可后要求 SARS-CoV-2 严重急性呼吸综合征冠状病毒 2 VRBPAC 疫苗和相关生物制品咨询委员会 MIS-C 儿童多系统炎症综合征 PREA 儿科研究公平法案 PVP 药物警戒计划 VE 疫苗有效性 CDC 疾病控制和预防中心 VAERS 疫苗不良事件报告系统 VOC 值得关注的变体 VSD 疫苗安全数据链接 US 美国
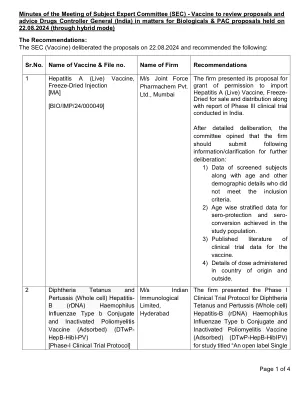
疫苗主题专家委员会 (SEC) 会议纪要,审查印度药品管理局关于生物制品问题的提案和建议
根据 2024 年 5 月 31 日 SEC 会议的建议,该公司提出了修订后的减毒活水痘疫苗 III 期临床试验方案,研究主题为“一项 III 期、多中心、随机、观察者盲法、活性对照、平行组、非劣效性研究,以评估水痘疫苗 (BARYCELA) 在 12 个月至 12 岁健康儿童人群中的免疫原性和安全性”。

2025 处方集(涵盖药物清单或“药物清单”) - MHBP
• 其他变更。我们可能会做出其他变更,这些变更会影响目前正在服用药物的会员。例如,我们可能会添加仿制药来替代目前在处方集中的品牌药,或添加新的生物仿制药来替代目前在处方集中的原始生物制品,或在添加相应药物后添加新的限制或将我们保留在处方集中的药物移至更高的费用分摊层级,或两者兼而有之。我们可能会在添加仿制药时从处方集中移除品牌药,或在添加生物仿制药时移除原始生物制品。我们还可能对品牌药或原始生物制品施加新的限制,或将其移至不同的费用分摊层级,或两者兼而有之。或者,我们可能会根据新的临床指南做出变更。

2025 处方集 - MHBP
• 其他变更。我们可能会做出其他变更,这些变更会影响目前正在服用药物的会员。例如,我们可能会添加仿制药来替代目前在处方集中的品牌药,或添加新的生物仿制药来替代目前在处方集中的原始生物制品,或在添加相应药物后添加新的限制或将我们保留在处方集中的药物移至更高的费用分摊层级,或两者兼而有之。我们可能会在添加仿制药时从处方集中移除品牌药,或在添加生物仿制药时移除原始生物制品。我们还可能对品牌药或原始生物制品施加新的限制,或将其移至不同的费用分摊层级,或两者兼而有之。或者,我们可能会根据新的临床指南做出变更。

CDER 2024 指导议程新的、修订草案和立即生效的指导 | 2024 年 7 月更新
• 加速药品和生物制品的审批 • 解决有关医疗器械和处方药的错误信息:问题与解答 2 • 未能满足加速上市后要求的民事罚款 • 首个可互换生物仿制药产品的独占性 • 机构审查委员会 (IRB) 对个体患者扩大临床试验药品和生物制品准入请求的审查 • 知情同意中的关键信息和促进理解 3 • 人用药品(包括生物制品)的 NDC 创建、分配、列出和适当使用 • 优先审查券计划 • 合格传染病产品指定——问题与解答 • 人用药品的重新包装和重新贴标:贴标;注册和列出、安全报告、供应链安全和良好生产规范 • 在药品 CGMP 类别结束时回应 FDA 483 号表格的观察结果 – 生物仿制药

2023 年 3 月 1 日 无标题信件 - Thomas Advanced Medical LLC
@thomasadvancedmedical.com 尊敬的 Angeles 女士和 Bogaart 博士: 美国食品药品管理局 (FDA) 生物制品评估与研究中心 (CBER) 合规与生物制品质量办公室审查了贵公司的网站 thomasadvancedmedical.com 以及下述其他在线资源和 FDA 可获得的其他信息。根据审查的材料,你和你的公司向消费者销售源自人类脐带、脐带血或羊膜组织的产品,你将它们称为“再生疗法解决方案”或“干细胞衍生生物制品”(统称为“你的产品”)。你和你的公司销售的产品用于治疗各种疾病或病症,包括一些严重或危及生命的疾病或病症,例如心脏病、阿尔茨海默病、帕金森病、肺部疾病和糖尿病。根据 FDA 审查的材料,您的产品适用于静脉注射、鞘内注射、关节内注射、皮下注射或肌肉注射或局部使用。根据您的网站: