XiaoMi-AI文件搜索系统
World File Search System生物制品

生物仿制药:十年经验和未来方向改善生物仿制药采用的策略和临床药理学的潜在作用
Yow-Ming Wang,博士 美国食品药品管理局临床药理学办公室生物仿制药和治疗性生物制品副主任 Yow-Ming Wang 博士目前担任 FDA 临床药理学办公室生物仿制药和治疗性生物制品副主任。她领导的治疗性生物制品项目旨在通过制定明确的政策、提高审查质量、促进知识共享、建立合作与拓展,促进生物产品开发中的科学和监管卓越性。 Gary Lyman,医学博士,公共卫生硕士,FACP,FRCP(爱丁堡),FASCO 教授;弗雷德哈钦森癌症研究中心公共卫生科学部和临床研究部癌症预防项目高级主管;哈钦森癌症结果研究所卫生保健质量与政策教授和兼职教授;华盛顿大学和杜克大学医学院公共卫生与药学系 Lyman 博士是弗雷德哈钦森癌症研究中心公共卫生科学与临床研究教授,同时还是哈钦森癌症结果研究所医疗质量与政策高级主管。他还是华盛顿大学和杜克大学医学院医学教授和公共卫生与药学副教授。

Align DUALPartnership (HMO D-SNP) 2025 年承保药物处方清单
其他变更。我们可能会做出其他变更,这些变更会影响目前服用药物的会员。例如,我们可能会添加一种新的仿制药来替代目前在处方集中的品牌药,或添加一种新的生物仿制药来替代目前在处方集中的原始生物制品,或添加新的限制或将我们保留在处方集中的药物移至更高的分摊等级或两者兼而有之。在我们添加相应的药物后,我们可能会在添加仿制药时从处方集中移除品牌药,或在添加生物仿制药时移除原始生物制品。我们还可能对品牌药或原始生物制品施加新的限制或将其移至不同的分摊等级,或两者兼而有之。我们可能会根据新的临床指南做出变更。如果我们从处方集中移除药物,对药物添加事先授权、数量限制和/或分步治疗限制或将药物移至更高的分摊等级,我们必须在变更生效前至少 30 天通知受影响的会员。或者,当会员要求补充药品时,他们可能会收到 31 天的药品供应量和变更通知。

SOPP 8217:新药临床试验申请行政处理和审查管理程序 版本:5 生效日期:2017 年 7 月 5 日
答:联邦食品、药品和化妆品法案 (FD&C Act) 第 505 条规定,药品或生物制品必须获得食品和药物管理局 (FDA) 的上市批准,才能跨州运输或分销。IND 是赞助商向 FDA 提出的豁免此法律要求的请求。IND 用于临床研究,以收集安全性和有效性信息,以支持生物制品和药品的上市申请或医学研究。根据 21 CFR 312 子部分 I 中的扩展获取条款,当没有可比或令人满意的替代治疗方案时,IND 还用于在临床试验之外治疗患有立即危及生命的疾病或严重疾病或病症的患者。
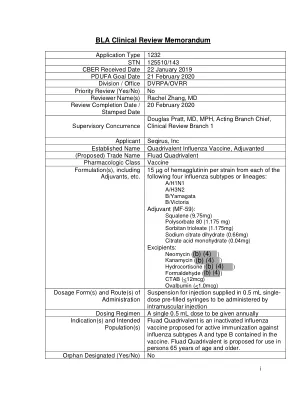
四价氟化物
AE 不良事件 AESI 特别关注的不良事件 aQIV 佐剂四价流感疫苗 AR 不良反应 aTIV 佐剂三价流感疫苗 BIMO CBER 生物研究监测 BLA 生物制品许可证申请 CBER 生物制品评价与研究中心 CFR 联邦法规 CI 置信区间 CMC 化学、制造与控制 CRF 病例报告表 CSR 临床研究报告 FAS 完整分析集 FDA 食品药品管理局 GMT 几何平均滴度 HA 血凝素 HI 血凝抑制 ICH 国际协调会议 ILI 流感样疾病 LL 下限 MedDRA 监管活动医学词典 NOCD 新发慢性病 OBE 生物统计学和流行病学办公室 OVRR 疫苗研究与审查办公室 PeRC 儿科审查委员会 PI 说明书 PMC 上市后承诺 PMR 上市后要求 PPS 按照方案集PREA 儿科研究公平法案 PT 首选术语 QIV 四价流感疫苗 RT-PCR 逆转录聚合酶链反应 SAE 严重不良事件 sBLA 补充生物制品许可申请 SCR 血清转化率 SOC 系统器官分类 STN 提交追踪编号 US 美国 WHO 世界卫生组织

标题 21 生物学家 12312024
合规和生物制品质量办公室 (OCBQ) 生物标准和质量控制部 (DBSQC) 生物化学、病毒学和免疫化学实验室分支 (LBVI) 摘要:食品药品管理局是监管、科学、公共卫生和消费者保护机构,负责确保所有人用和动物药物以及医疗器械安全有效;化妆品、食品、食品添加剂、食用动物的药物和药用饲料以及辐射发射设备安全;并且在美国销售的所有此类产品均有充分、真实和信息丰富的标签,并安全和正确地储存、运输、制造、包装和监管。生物制品评估和研究中心 (CBER) 的使命是通过监管生物和相关产品(包括血液、疫苗、过敏原、组织以及细胞和基因疗法)来保护和增进公众健康。概述:

招聘工程和物理科学 DTP 博士生
能源与生物制品研究所 - EBRI 塑料废物、化学回收、循环经济、催化热解、可持续性、生物质、人工智能、氢、膜、燃料电池、生物燃料、非均相催化、膜、水处理、废水、化学工程、二氧化碳转化、清洁能源、绿色催化、可持续塑料、生物加工、废物价值化、微生物学、生物能源、气化、生物炭、封存、热存储;热升级;生物热;可再生热能、机械工程、热能、加热和冷却、空调、热泵、平台化学、水相、催化、催化剂开发(合成、特性和测试)、纳米材料、低碳燃料、先进燃烧、排放、发动机性能 请参阅能源与生物制品研究所 - EBRI 的列表,项目在这里

FDARA 分子靶向肿瘤药物儿科研究实施指南:FD&C 法案第 505B 条修正案 - 行业指南
1 本指南由美国食品药品管理局肿瘤卓越中心与药品评估与研究中心以及生物制品评估与研究中心合作制定。 2 在本指南中,药品和药品产品包括根据《联邦食品药品和化妆品法案》(FD&C 法案)第 505 节批准的药品(21 USC 355)和根据《公共卫生服务法案》第 351 节许可的生物制品(42 USC 262)。 3 有关 PREA 要求和拟议生物仿制药产品的更多信息,请参阅行业指南草案《生物仿制药开发和 BPCI 法案新修订问答草案》(修订版 2)(2018 年 12 月)中的 QI16。(最终版本将代表 FDA 目前对此主题的看法)。 4 公法 115-52、131 Stat. 1005(2017 年 8 月 18 日)。

评估 COVID-19 预防或治疗药物和生物制品临床试验中成人和青少年门诊受试者的 COVID-19 相关症状
本指南为申办方和研究者提供了在评估用于预防或治疗门诊成人和青少年受试者的 COVID-19 的药物或生物制品的临床试验中如何测量和分析与 2019 冠状病毒病 (COVID-19) 相关的常见症状的方法的考虑。本指南不适用于评估用于治疗或预防儿童和成人感染后 COVID-19 状况(例如,长期 COVID、多系统炎症综合征)的产品的开发计划,也不适用于预防性疫苗的开发计划。本指南不涉及临床试验设计方面的考虑因素,除了涉及测量和分析门诊患者 COVID-19 相关症状的考虑因素之外。需要住院治疗的 COVID-19 患者的考虑因素不在本指南的范围内。

FDA AI ML 讨论文件_V2
Flatiron 对 FDA 的讨论文件“人工智能和机器学习在药品和生物制品开发中的应用”(“讨论文件”)表示赞赏和欢迎。1 我们认识到人工智能/机器学习具有通过创造巨大效率来促进药品和生物制品开发的潜力,而这种效率的提高部分得益于数据收集和证据生成方面的快速技术创新。我们还认识到需要仔细评估这项技术是否会带来特定的风险或危害。Flatiron 支持 FDA 的使命,即确保这些创新的全部益处得以实施并造福公众。因此,我们赞赏该机构通过讨论文件主动与利益相关者展开对话,并计划就这个跨多个部门的快速发展话题举行未来多利益相关者研讨会,以促进相互学习和讨论并塑造监管格局。

行业指南
I. 引言 本指南提供建议,以协助业界和其他参与开发具有细胞毒性小分子药物或有效载荷的抗体-药物偶联物 (ADC) 的各方。具体而言,本指南解决了 FDA 当前关于临床药理学考虑因素的想法,以及关于生物分析方法、给药策略、剂量和暴露反应分析、内在因素、QTc 评估、免疫原性和药物间相互作用 (DDI) 的建议。本指南中讨论的原则可能不适用于开发其他类型的 ADC(例如,具有除细胞毒性小分子药物以外的有效载荷和/或用于除肿瘤学以外的适应症的 ADC)。本指南专门概述了 ADC 开发计划的临床药理学考虑因素,并在适当的时候引用其他相关指南。2 ADC 受所有相关生物制品法律法规的约束,包括 PHS 法案第 351 节(42 USC 262)中概述的产品开发、测试和批准法律法规。鉴于 ADC 包括小分子药物 3,还有其他适用于 ADC 的指导原则,而这些指导原则不一定适用于其他生物制品。值得注意的是,本指导原则并不侧重于任何特定 ADC 的开发,有关特定 ADC 的监管建议和开发计划的问题应向相应的 FDA 审查部门提出。此外,对于临床和非临床数据,所谓的“独立”申请(例如,根据 FDA 提交的生物制品许可申请 (BLA))的申请人