XiaoMi-AI文件搜索系统
World File Search System肌萎缩

腓骨肌萎缩症:从巴西的历史地标到当前的护理观点
摘要 遗传性运动和感觉神经病,又称腓骨肌萎缩症 (CMT),传统上是指一组以神经病为主要或唯一特征的遗传性疾病。其患病率因研究人群不同而异,估计在 1:2,500 至 1:10,000 之间。自 1989 年 Vance 等人在 17 号染色体上发现 PMP22 基因重复以来,已有 100 多个基因与这组疾病有关,我们在患者护理方面取得了进展,发现了相关疾病并采取了更好的支持性治疗,包括临床和外科干预。此外,随着遗传学领域的发现,包括 RNA 干扰和基因编辑技术,新的治疗前景开始出现。在目前的工作中,我们报告了巴西 CMT 研究方面最重要的里程碑,并对诸如我们人口中与 CMT 相关的不同基因的频率、疼痛的流行率、对怀孕的影响、呼吸特征以及新疗法的开发等主题进行了全面的回顾。

38在γ-突触核蛋白中发现了两名肌萎缩症患者...
了解突触核素蛋白在体外和细胞中如何形成淀粉样蛋白如何对了解疾病至关重要。先前的研究表明,α-突触核蛋白的P1区(残基36-42)控制淀粉样蛋白的形成。我们在这里报告了在两个患有肌萎缩性侧面硬化症的个体中发现的γ-突触蛋白(γSyn)(Met38至Ile)的P1区域中的单个核苷酸多态性。两个个体在同一基因(glu110 to val)中都有第二个多态性,通常在普通人群中发现。我们表明,ILE38-含有γ静态形式的淀粉样蛋白在体外快速淀粉样蛋白,而Met38并未聚集成淀粉样蛋白,而Val110具有保护性,从而减慢了聚集。结果突出了P1序列在蛋白质淀粉样蛋白倾向之间平衡的关键作用。

Taldefgrobep Alfa 和脊髓性肌萎缩症 3 期 RESILIENT 试验
摘要:脊髓性肌萎缩症 (SMA) 是一种罕见的遗传性神经退行性疾病,由存活运动神经元 (SMN) 蛋白生成不足引起。SMN 蛋白水平降低会导致运动神经元丢失,从而引起肌肉萎缩和虚弱,损害日常功能并降低生活质量。SMN 上调剂可改善 SMA 患者的临床状况并提高其存活率,但仍存在大量未满足的需求。肌生长抑制素是一种与激活素 II 受体结合的 TGF-β 超家族信号分子,可负向调节肌肉生长;肌生长抑制素抑制是一种有前途的增强肌肉的治疗策略。将肌生长抑制素抑制与 SMN 上调相结合是一种针对整个运动单元的综合治疗策略,为 SMA 带来了希望。Taldefgrobep alfa 是一种新型的全人源重组蛋白,可选择性地与肌生长抑制素结合并竞争性地抑制通过激活素 II 受体发出信号的其他配体。鉴于 taldefgrobep 在神经肌肉疾病患者中具有可靠的科学和临床依据以及良好的安全性,RESILIENT 3 期随机安慰剂对照试验正在研究 taldefgrobep 作为 SMA 中 SMN 上调剂的辅助剂 (NCT05337553)。本文回顾了肌生长抑制素在肌肉中的作用,探讨了 taldefgrobep 的临床前和临床开发,并介绍了 taldefgrobep 在 SMA 中的 3 期 RESILIENT 试验。
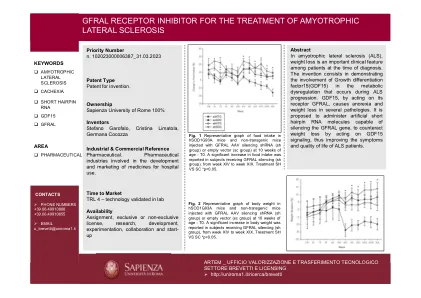
用于治疗肌萎缩侧索硬化症的 GFRAL 受体抑制剂
技术与优势 ALS 患者的发病率为 56%-62%,体重减轻被定义为一个重要且独立的预后因素。此外,多项研究报告称,疾病进展与诊断时的体重减轻或低体重指数 (BMI) 成正比。通过实施特定的饮食计划,包括各种高热量脂肪或糖饮食,ALS 患者的疾病进展速度减慢,生活质量提高。这一观察结果关注的是 ALS 患者的代谢状况对疾病进展的影响。GDF15 细胞因子受体多核苷酸抑制剂 GFRAL 旨在改善 SLA 患者的体重减轻和恶病质。

肌萎缩侧索硬化症等罕见神经退行性疾病行动计划
加速获得 ALS 关键疗法法案 2021 年 12 月 23 日,总统签署了《加速获得 ALS 关键疗法法案》3(以下简称“ALS 法案”),该法案指示美国卫生与公众服务部 (HHS) 采取一系列行动,包括根据 ALS 法案第 4 条的要求,制定一项行动计划,以促进 ALS 和其他罕见神经退行性疾病药物的开发和获取。具体而言,第 4 条指出,在 ALS 法案颁布后六个月内,FDA 应在其网站上发布一份行动计划,描述 FDA 打算在计划发布后的五年内就计划增强、政策制定、监管科学举措和其他适当举措采取的行动,以便:

全基因组鉴定肌萎缩侧索硬化症的遗传基础
张赛, 1 , 8 Johnathan Cooper-Knock, 2 , 8 Annika K. Weimer, 1 Minyi Shi, 1 Tobias Moll, 2 Jack NG Marshall, 2 Calum Harvey, 2 Helia Ghahremani Nezhad, 2 John Franklin, 2 Cleide dos Santos Souza, 2 Ke Ning, 2 Cheng Wang, 3 Jingjing Li, 3 Allison A. Diliot, 4 Sali Farhan, 4 Eran Elhaik, 5 Iris Pasniceanu, 2 Matthew R. Livesey, 2 Chen Eitan, 6 Eran Hornstein, 6 Kevin P. Kenna, 7 Project MinE ALS 测序联盟, Jan H. Veldink, 7 Laura Ferraiuolo, 2 Pamela J. Shaw, 2 和 Michael P. Snyder 1 , 9 , * 1 遗传学系中心斯坦福大学医学院基因组学和个性化医学系,斯坦福,CA 94305,美国 2 谢菲尔德大学谢菲尔德转化神经科学研究所,谢菲尔德,S10 2HQ,英国 3 伊莱和埃迪丝布罗德再生医学和干细胞研究中心、巴卡尔计算健康科学研究所、帕克癌症免疫治疗研究所和加州大学旧金山分校医学院神经病学系,旧金山,CA 94143,美国 4 麦吉尔大学蒙特利尔神经病学研究所神经病学和神经外科系,蒙特利尔,QC H3A 1A1,加拿大 5 隆德大学生物系,隆德 223 62,瑞典 6 魏茨曼科学研究所分子遗传学系,雷霍沃特 7610001,以色列 7 大学医学中心鲁道夫马格努斯脑中心神经病学系乌得勒支,乌得勒支 3584 CX,荷兰 8 这些作者贡献相同 9 主要联系人 *通信地址:mpsnyder@stanford.edu https://doi.org/10.1016/j.neuron.2021.12.019

肌萎缩侧索硬化症 (ALS) 的实验医学成功之路 (EXPERTS-ALS)
简明英语摘要 背景和研究目的 肌萎缩侧索硬化症 (ALS) 是最常见的运动神经元疾病 (MND),是一种影响 1/300 人的致命疾病。ALS 会导致肌肉萎缩,并随着时间的推移而恶化,导致失去行走、说话、进食和最终呼吸的能力。ALS 没有治愈方法或高效的减缓疾病的疗法。从历史上看,ALS 的药物试验一直没有成功。人们认为,一种可以在 ALS 患者而不是细胞或动物模型中快速筛选药物的过程将更好地为所需的大型 III 期试验选择治疗方法。EXPERTS-ALS 是一项英国多中心研究,它将通过观察血液测试标记物的变化来筛选药物是否有可能减缓 ALS 的进展。血液中一种称为神经丝轻链 (NFL) 的标记物(神经细胞的组成部分)的水平越高,ALS 致残率就越快。如果发现血液中的 NFL 水平出现大幅下降,就会建议将药物推向 III 期试验。

Janus 激酶抑制剂是治疗肌萎缩侧索硬化症的潜在药物
© 作者 2023。开放存取 本文根据知识共享署名 4.0 国际许可协议进行授权,允许以任何媒体或格式使用、共享、改编、分发和复制,只要您给予原作者和来源适当的信任,提供知识共享许可协议的链接,并指明是否做出了更改。 本文中的图片或其他第三方资料包含在文章的知识共享许可协议中,除非资料的致谢中另有说明。 如果资料未包含在文章的知识共享许可协议中,且您的预期用途不被法定规定允许或超出了允许的用途,则需要直接从版权所有者处获得许可。 要查看此许可证的副本,请访问 http://creat iveco mmons. org/licen ses/ by/4. 0/。知识共享公共领域贡献豁免(http://creat iveco mmons. org/publi cdomina/zero/1. 0/)适用于本文中提供的数据,除非数据版权声明中另有说明。


深入探索进展:脊髓性肌萎缩症当前治疗进展综述
脊髓性肌萎缩症 (SMA) 是一种罕见疾病,与基因有关,其特征是肌肉逐渐衰弱和退化,常常导致严重残疾和过早死亡。在过去十年中,SMA 治疗领域取得了显著进展,彻底改变了患者护理的格局。一项关键进展是基因靶向疗法的开发,例如 nusinersen、onasemnogene abeparvovec 和 risdiplam,它们在减缓疾病进展方面表现出前所未有的功效。这些疗法旨在通过靶向存活运动神经元 (SMN) 基因来解决 SMA 的根本原因,有效恢复缺陷的 SMN 蛋白水平。这些创新方法的出现改变了许多 SMA 患者的预后,为曾经治疗手段有限的患者带来了一线希望。此外,小分子化合物和 RNA 靶向策略的出现扩大了针对 SMA 的治疗手段。这些新干预措施表现出多种作用机制,包括 SMN 蛋白稳定和 RNA 剪接调节,展现了 SMA 治疗研究的多面性。制药行业、研究中心和患者权益团体的共同努力在加速将科学发现转化为可见的临床效益方面发挥了重要作用。这篇评论不仅突出了 SMA 治疗取得的显著进展,还为持续努力提高可及性、优化治疗策略、康复(护理和疗法)以及最终为改善 SMA 患者的生活质量铺平道路带来了希望之光。