XiaoMi-AI文件搜索系统
World File Search System遗传病

研究科学家 / 高级研究科学家 – 新平台
Tevard Biosciences 是 mRNA 调节疗法的先驱,用于治疗多种遗传疾病。这家私营生物技术公司由麻省理工学院教授兼怀特黑德研究所创始成员 Harvey Lodish 与生命科学企业家兼高管 Daniel Fischer 和 Warren Lammert(罕见遗传病儿童的父亲)以及科学联合创始人 Jeff Coller(约翰霍普金斯大学医学院分子生物学和遗传学系彭博杰出教授)共同创立。Tevard 正在探索其新型抑制 tRNA、增强 tRNA 和 mRNA 调节平台在神经系统疾病、心脏病和肌营养不良症中的应用。职位摘要:我们正在寻找一位在分子/细胞生物学方面经验丰富的高技能科学家加入 Tevard 团队。成功候选人将在 Tevard 新型治疗平台的设计和开发中发挥主导作用。理想的候选人应具有丰富的分子/细胞生物学、体外或体内疾病模型(骨骼肌或心肌细胞优先)经验,并使用最先进的技术设计和领导研究。该职位要求具有创新思维、高度注重细节以及协作解决难题的能力。该职位的主要职责包括:

pr_amp_tmb_testing_guideli ...
由AMP,ASCO,CAP和M.D.- 2024年6月6日 - 全球全球分子诊断专业社会的分子病理学协会(AMP)今天发表了一系列基于证据的建议,用于对肿瘤突变负担(TMB)测试作为潜在预测性生物标记物的分析验证和报告,用于免疫检查点侵入点(ICI)的潜在预测生物标志物。这些建议包括TMB分析的分析前,分析和分析后因素,并强调出版物中全面的方法论描述的重要性以允许分析之间的可比性。手稿,“肿瘤突变负担分析验证和报告的建议:分子病理协会,美国病理学家学院和癌症免疫疗法协会的共同共识建议”,在《分子诊断杂志》发表之前在线发布。ICI疗法已转化了患有多种癌症患者的一部分患者护理。结果,对预测性生物标志物(例如TMB)仍然具有重大兴趣,这些生物标志物可以识别患者从这些治疗中受益更可能受益。但是,TMB的计算,报告和解释可能在不同的实验室之间有所不同。建立了AMP TMB工作组,以评估现有的实验室实践,并为临床TMB测试的分析验证和报告制定基于证据的标准。我们的成员是这些建议旨在作为基于科学文献,观察性调查数据以及工作组主题专家的专业经验的参考指南。“虽然TMB已成为ICI治疗的潜在预测生物标志物,计算和报告TMB的多种方法,以及关于临床测定验证的全面方法论描述,对采用构成了重大挑战,” Larissa V. Furtado,AMP TMB TMB TMB Working Groups和Morecull Pathist in St. jude at Set. judet。LarissaV. Furtado说。“这份新报告不仅总结了与TMB测试相关的现有知识和挑战,而且还提供了有关如何在临床环境中最好地验证和报告这些测定法的共识建议。” “AMP is committed to helping laboratories overcome challenges and improve current test and interpretation practices,” said Susan Hsiao, MD, PhD, Chair of the 2024 AMP Clinical Practice Committee, Member of the AMP TMB Working Group, and Associate Professor of Pathology and Cell Biology at Columbia University Vagelos College of Physicians and Surgeons.“与我们所有基于证据的指南一样,随着新的技术和科学进步的可用,AMP临床实践委员会将继续重新评估和修改这些TMB建议。”要阅读完整的手稿,请访问https://doi.org/10.1016/j.jmoldx.2024.05.002。关于AMP,分子病理协会(AMP)成立于1995年,为分子诊断的新兴领域提供结构和领导。AMP的2,900多名成员练习分子诊断的各种学科,包括生物信息学,传染病,遗传病和肿瘤学。AMP的2,900多名成员练习分子诊断的各种学科,包括生物信息学,传染病,遗传病和肿瘤学。

四例谷胱甘肽合成酶缺乏症患者的基因型和临床表型
背景和目的:谷胱甘肽合成酶缺乏症 (GSSD) 是一种常染色体隐性遗传病,文献中描述了约 80 名患者。目前,人们对 GSSD 的基因型-表型相关性知之甚少,尽管可以通过突变分析在一定程度上预测其严重程度。在这里,我们描述了四名患有 GSSD 的患者,并评估了他们的基因型和表型。此外,我们还提供了最新的文献综述。方法:我们回顾性地审查了巴勒斯坦耶路撒冷 Al-Makassed 医院过去十年中所有患有 GSSD 患者的病历。我们回顾了医疗管理的文献和最新的治疗研究,并讨论了表型-基因型相关性。结果:我们描述了四名确诊为 GSSD 的患者。临床表现的严重程度各不相同,但患者通常表现为溶血性贫血和乳酸性酸中毒。尿液有机酸分析显示大量乳酸和焦谷氨酸排泄。所有患者均接受了 N-乙酰半胱氨酸、维生素 E、维生素 C 和碳酸氢钠治疗。除一名患者在两个月大时死亡外,所有患者在治疗后均有显著改善。结论:GSSD 的表现与许多其他疾病相似,有时会导致诊断延迟。早期开始治疗可以改善临床结果和整体发展。如果高度怀疑患有 GSSD,则重要的是考虑进行 mRNA 测序,以防止在存在剪接位点突变时延误诊断。

视网膜纤毛病和潜在的基因疗法
摘要:人类感光细胞的功能依赖于高度特化的纤毛。纤毛功能的紊乱通常会导致感光细胞死亡和视力丧失。视网膜纤毛病是一种遗传多样性的视网膜遗传病,会影响感光细胞纤毛的各个方面。尽管利用动物疾病模型对视网膜纤毛病的理解取得了进展,但它们往往无法准确模拟观察到的患者表型,这可能是由于结构和功能与人类视网膜存在偏差。人类诱导多能干细胞 (hiPSC) 可用于生成替代疾病模型,即 3D 视网膜类器官,其中包含所有主要的视网膜细胞类型,包括带有纤毛结构的感光细胞。这些视网膜类器官有助于研究人类衍生系统中的疾病机制和潜在疗法。三维视网膜类器官仍是一项发展中的技术,尽管取得了令人瞩目的进展,但仍存在一些局限性。本综述将讨论 hiPSC 衍生的视网膜类器官技术现状,该技术可准确模拟与基因(包括 RPGR 、 CEP290 、 MYO7A 和 USH2A )相关的主要视网膜纤毛病。此外,我们还将讨论针对视网膜纤毛病的新型基因治疗方法的开发,包括大基因的传递和基因编辑技术。

从 CRISPR 靶向 RNA 编辑中获得的见解......
摘要:成簇的规律间隔短回文重复序列(CRISPR)/CRISPR相关蛋白(Cas)系统,尤其是II型(Cas9)系统,在DNA打靶方面得到了广泛的发展,形成了一套成熟的精准基因编辑系统。但CRISPR-Cas系统在RNA上的基础研究和应用尚处于早期阶段。近期,CRISPR-Cas13 VI型系统的发现,为拓展RNA打靶技术提供了可能,具有广阔的应用前景。大多数VI型Cas13效应子具有双核酸酶活性,能催化前crRNA转化为成熟的crRNA,并产生较强的RNA切割活性。Cas13能特异性识别靶向RNA片段,激活Cas13/crRNA复合物进行侧切活性。 Cas13X蛋白是Cas13家族中最小的效应子,长度为775个氨基酸,由于其不受前间隔区侧翼序列(PFS)限制、易于包装、不造成永久性损伤,是一种很有前途的RNA靶向平台。本研究重点介绍了CRISPR-Cas13家族靶向RNA编辑的最新进展,并讨论了Cas13在基础研究、核酸诊断、核酸追踪和遗传病治疗中的应用。此外,我们阐明了Cas13蛋白家族的结构及其分子机制,并提出了CRISPR-Cas13家族靶向RNA编辑的未来愿景。

混合方法研究身体和情感自我...
抽象的多囊卵巢综合征(PCOS)是一种罕见的遗传病,影响数百万的生殖年龄妇女。月经不规则,多毛,痤疮,脱发,肥胖和不育都是PCOS的症状,这是由雄激素水平高于正常的症状引起的。此外,月经不规则性是PCOS的普遍症状,通常是青少年中的第一个临床表现。这项研究确定了物理和情感自我概念的构造,以越来越多地影响PCOS的青少年,因为这些临床表现的负面影响会影响他们对自己的看法和信念,这会改变他们的自我概念或对身体自我的看法。此外,它影响了他们的情感自我概念,从而影响了他们对自己的感觉。在这项研究中,基于正念的压力减轻计划(MBSR)在线促进,以提高使用顺序解释性混合方法研究设计的16名被诊断为多囊卵巢综合征的青少年的身体和情感自我概念的水平。多维自我概念量表(MSC)和单个访谈已纳入数据。结果清楚地证明,在MBSR促进之前和之后,身体和情感自我概念的得分显着差异。这项研究证实,MBSR通过PCOS的身体和情感自我概念增强了青少年。关键字:与多囊卵巢综合征(PCOS)简介使用主题分析分析了MBSR促进之前和之后产生的主题,该主题分析进一步建立了发现,以解决有关该主题的研究的缺乏,并作为MBSR功效的基础。
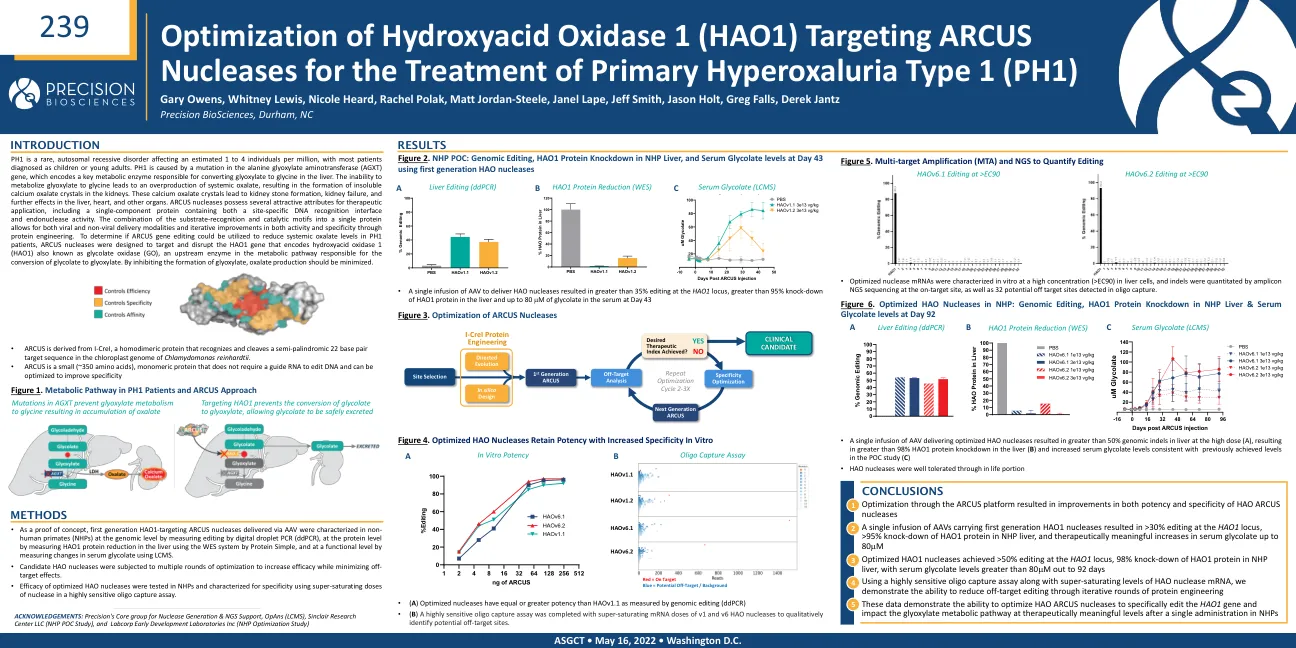
针对ARCUS核酸酶的羟基氧化酶1(HAO1)的优化用于治疗原发性高草酸尿症1型(PH1)
PH1 是一种罕见的常染色体隐性遗传病,每百万人中估计有 1 至 4 人患有此病,大多数患者在确诊时为儿童或年轻人。PH1 是由丙氨酸乙醛酸转氨酶 (AGXT) 基因突变引起的,该基因编码一种关键代谢酶,负责在肝脏中将乙醛酸转化为甘氨酸。无法将乙醛酸代谢为甘氨酸会导致全身性草酸过量产生,从而导致肾脏中形成不溶性草酸钙晶体。这些草酸钙晶体会导致肾结石形成、肾衰竭,并进一步影响肝脏、心脏和其他器官。ARCUS 核酸酶具有多种有利于治疗应用的特性,包括一种单组分蛋白质,既包含位点特异性 DNA 识别界面,又包含核酸内切酶活性。将底物识别和催化基序组合成单一蛋白质,既可用于病毒传递方式,也可用于非病毒传递方式,并通过蛋白质工程不断提高活性和特异性。为了确定 ARCUS 基因编辑是否可用于降低 PH1 患者的全身草酸水平,ARCUS 核酸酶被设计用于靶向和破坏编码羟基酸氧化酶 1 (HAO1) 的 HAO1 基因,HAO1 也称为乙醇酸氧化酶 (GO),是代谢途径中负责将乙醇酸转化为乙醛酸的上游酶。通过抑制乙醛酸的形成,草酸的产生应被最小化。

全基因组分析揭示了非...的贡献。
摘要背景:逆转录转座子被认为是孟德尔遗传病的病因,但它们在自闭症谱系障碍 (ASD) 中的作用尚未系统地定义,因为只有从全基因组测序 (WGS) 数据中才能以足够的灵敏度调用它们,并且最近才有足够大的队列进行此项分析。结果:我们通过建立可扩展的计算流程来检测逆转录转座子插入,分析了来自 Simons Simplex Collection 的 2288 个 ASD 家族的 WGS 数据。我们报告了 86,154 个多态性逆转录转座子插入(其中 > 60% 以前未报告)和 158 个新生逆转录转座事件。ASD 个体和未受影响的兄弟姐妹之间新生事件的总体负担相似,Alu、L1 和 SVA 分别每 29、117 和 206 个出生发生 1 个新生插入,总共每 21 个出生发生 1 个新生插入。然而,ASD 病例显示 ASD 基因中 L1 从头插入比预期更多。此外,我们仅在 ASD 个体中观察到功能丧失不耐受基因中的外显子插入,包括 CSDE1 中可能致病的外显子插入。结论:这些发现表明内含子和外显子逆转录转座子插入对 ASD 的影响不大但很重要,表明了 WGS 对其分析的重要性,并强调了特定生物信息学工具对高通量检测逆转录转座子插入的实用性。关键词:转座因子、逆转录转座子、自闭症谱系障碍、从头插入、多态性插入、从头率、Alu、SVA、LINE-1、神经生物学

意大利在罕见疾病 CRISPR 试验中取得突破
基因治疗的发展以及目前有利的成簇规律间隔短回文重复序列 (CRISPR) 方法使得多项临床试验得以实施,旨在研究基因治疗对罕见疾病的可能疗效。罕见疾病对全球构成挑战,因为它们对卫生系统的总体影响巨大,而它们个别的罕见发生阻碍了有效疗法的研究和开发。尽管个别罕见疾病的患病率很低,但已确定的罕见疾病超过 7,000 种,影响着全球 3.5-5.9% 的人口。罕见疾病大多是慢性的,约 80% 是由早期发病的基因突变引起的。在意大利,2021 年记录的罕见疾病患者超过 400,000 人。由于地理位置和历史原因,意大利在两种罕见遗传病的存在和流行方面有着令人遗憾的统计数据,即β-地中海贫血,全球约有 9000 万携带者,其中 40 万人实际受到影响,以及镰状细胞病,全球约有 3 亿携带者,650 万人受到影响。基因组研究的进步使意大利能够加入临床试验,研究有效和有效的 BT 和 SCD 基因疗法。本研究报告了罕见病在意大利的影响、正在进行的研究、使用 CRISPR 方法进行 BT 和 SCD 试验的最新成果以及 CRISPR 技术应用于罕见病的剩余障碍,同时也概述了罕见病基因治疗的最新挑战和未来机遇。

儿童中脊柱肌肉萎缩2型的病例
抽象的脊柱肌肉萎缩(SMA)是一种神经退行性疾病,影响了10,000个活产中4个。这是一种由位于5q染色体的生存运动神经元基因1(SMN1)突变引起的常染色体隐性遗传病。有五种类型的SMA,从类型0到类型4。取决于类型,SMA会导致严重的残疾和死亡。此案例报告提出了一个六个月大的男婴,其中有运动发育总额。没有机会在初级保健中发现此病例。该婴儿被私人儿科医生转介给政府医院的门诊儿科诊所,因为他失去了自己的能力,抬起臀部并没有支持。基因检测证实了SMA 2型的诊断。接受SMA儿童的诊断和照顾对父母和看护者来说是一项改变生活的事件。基因疗法的可用性可能会改变SMA患者的预后和预后,如果有可能提供。此案强调了孩子的疾病对家庭的影响以及多学科团队在管理SMA方面的重要性。初级保健医师在进行彻底的儿童健康监测方面发挥了关键作用,以确保早期识别并随着疾病的成年疾病的发展,为孩子和父母提供支持。这包括提供长期的社会心理支持以改善其生活质量。关键字:脊柱肌肉萎缩,2型,总体运动发育延迟,遗传