XiaoMi-AI文件搜索系统
World File Search SystemAngelman

rett综合征 - 基因和troinetide治疗中的Advans Rett综合征 - 基因治疗和troinetide的进展
RETT综合征是一种罕见的,严重的神经发育障碍,具有X连锁的主要遗传。 它主要影响妇女,由于婴儿期快速发育消退而导致认知和身体障碍。 Rett综合征通常在六到18个月大的儿童中被认可,当时他们开始错过发展里程碑或失去所获得的能力。 一种特征症状涉及连续重复的手动运动。 RETT综合征是女孩中最常见的复杂残疾原因之一。 但是,这种情况可能会被误诊。 应考虑的鉴别诊断包括脑瘫,自闭症,安吉尔曼综合症和非特异性发育延迟。 rett综合征与编码甲基-CPG结合蛋白2的基因的功能丧失突变有关(约占报告病例的90%)。 这些突变与影响神经元和轴突连接的发展有关。 自RETT综合征的第一个报告以来,在过去的50年中,取得了进展。 在实验室和临床环境中都在测试了一些有前途的临床试验和令人兴奋的新型治疗选择。 研究结果导致了2023年3月第一种治疗药物Trofinetide的注册。 在最近的研究中发现该药物可提高大脑功能和沟通技巧。 也有有希望的临床试验研究了突变基因的替代。RETT综合征是一种罕见的,严重的神经发育障碍,具有X连锁的主要遗传。它主要影响妇女,由于婴儿期快速发育消退而导致认知和身体障碍。Rett综合征通常在六到18个月大的儿童中被认可,当时他们开始错过发展里程碑或失去所获得的能力。一种特征症状涉及连续重复的手动运动。RETT综合征是女孩中最常见的复杂残疾原因之一。但是,这种情况可能会被误诊。应考虑的鉴别诊断包括脑瘫,自闭症,安吉尔曼综合症和非特异性发育延迟。rett综合征与编码甲基-CPG结合蛋白2的基因的功能丧失突变有关(约占报告病例的90%)。这些突变与影响神经元和轴突连接的发展有关。取得了进展。在实验室和临床环境中都在测试了一些有前途的临床试验和令人兴奋的新型治疗选择。研究结果导致了2023年3月第一种治疗药物Trofinetide的注册。在最近的研究中发现该药物可提高大脑功能和沟通技巧。也有有希望的临床试验研究了突变基因的替代。这项研究旨在分析RETT综合征中最新的药理学治疗和基因治疗,这给患者及其家人带来了希望,他们预见了Rett综合征是可逆且可治愈的疾病的未来。

雷特综合征 - 基因和曲芬奈肽治疗的进展 雷特综合征 - 基因和曲芬奈肽治疗的进展
雷特综合征是一种罕见的严重神经发育障碍,具有 X 连锁显性遗传。该病主要影响女性,由于婴儿期快速发育倒退而导致认知和身体障碍。雷特综合征通常在 6 至 18 个月大的儿童中发现,此时他们开始错过发育里程碑或失去已获得的能力。一种典型症状是持续重复的手部动作。雷特综合征是女孩复杂残疾的最常见原因之一。然而,这种疾病可能会被误诊。应考虑的鉴别诊断包括脑瘫、自闭症、天使综合征和非特异性发育迟缓。雷特综合征与编码甲基 CpG 结合蛋白 2 的基因的功能丧失突变有关(约占报告病例的 90%)。这些突变与影响神经元和轴突连接的发育有关。自首次报告雷特综合征以来,过去 50 年来取得了进展。实验室和临床环境中正在测试多项有希望的临床试验和令人兴奋的新型治疗方案。研究结果促使第一种治疗药物曲芬奈肽于 2023 年 3 月注册。最近的研究发现,这种药物可以改善大脑功能和沟通技巧。还有一些有希望的临床试验正在研究突变基因的替换。这项研究旨在分析雷特综合征的最新药物治疗和基因疗法,这给患者及其家属带来了一线希望,他们期待未来雷特综合征是一种可逆和可治愈的疾病。

整合强化学习和认真的游戏,以支持罕见的遗传疾病和神经发育障碍的人:父母和看护者的结果
个体正在努力应对罕见的遗传疾病,例如Angelman,Cornelia de Lange,脆弱的X和RETT综合征,在导航其日常环境时面临着巨大的挑战。除了智力障碍,沟通统计和感官障碍外,这些人还经常患有严重的运动障碍。这种复杂的情况不仅严重损害了他们的生活质量,而且对照料者和家庭造成了增加的负担(Krath等,2021)。为了应对这些挑战,技术干预已成为有前途的解决方案。认真的游戏并利用新技术,具有教育,诊断和康复目的的身临其境和娱乐性的体验。越来越多地采用了基于人工情报的计划,尤其是那些采用强化学习的程序。这种复杂的方法涉及一种人工智能的代理,与参与者的表现不断相互作用,以实时调整任务或活动的复杂性或困难(Krath等,2021; Liu等,2022)。这种个性化的适应性确保了最佳的用户参与度和效果。在本文中,我们主张将严肃的游戏和强化学习的整合到服务和康复目标。这种合并的方法可能提出了一种量身定制的解决方案,以促进患有罕见遗传疾病的个体的适应性反应。我们探索了各种领域,包括具有执行功能的认知技能,沟通能力和管理具有挑战性的行为。我们承认对参与者的生活质量的深刻影响,提供了说明性的例子来强调我们的观点。我们的创新方法将游戏融合与伯爵的认知发展理论相结合,将其分类为促进新的适应技巧的认知框架(Robb等,2019)。

针对TRKB – PSD-95耦合以减轻神经系统...
摘要tropomyosin受体激酶B(TRKB)信号传导在树突生长和树突状脊柱形成中起关键作用,以促进学习和记忆。突触下脑衍生的神经营养因子的活性依赖性释放与突触前或突触后TRKB结合,导致突触的增强,反映出长期增强。突触后,突触后密度蛋白-95与TRKB的缔合增强了磷脂酶Cγ-CA 2+ /钙调蛋白依赖蛋白依赖性蛋白激酶II和磷脂酰肌醇3-激酶机械靶标的雷帕木霉素信号的长期有效性所需。在这篇综述中,我们讨论了TRKB poStsynaptic蛋白-95耦合作为一种有前途的策略,以放大脑衍生的神经营养因子信号传导,以开发针对特定神经系统疾病的新型疗法。,并增强了突触后密度蛋白-95与TRKB信号的关联,可能会减轻精神分裂症和抑郁症神经连通性的缺乏。用脑源性神经营养因子的治疗是有问题的,这是由于药代动力学不良,脑穿透性低以及p75神经营养蛋白受体或截短的TRKB.T1同工型引起的副作用。尽管正在深入研究激活TRKB的TRKB激动剂和抗体,但它们无法区分多个人类TRKB剪接同工型或细胞类型特异性功能。靶向trkB - postsynaptic蛋白-95耦合耦合提供了一种替代方法,可在局部突触部位特异性提高TRKB信号传导与全球刺激,从而冒着许多不良副作用的风险。关键词:Angelman综合征;自闭症;脑衍生的神经营养因子;沮丧;神经退行性疾病;神经发育障碍;突触后密度蛋白-95;突触可塑性; trkb

ube3a:自闭症谱系障碍(ASD)的作用和生物标志物研究的潜在候选者和设计治疗策略
摘要:CDC自闭症和发展障碍监测网络的发布报告表明,估计每44(2.3%)8岁儿童中有1个(2.3%)在2018年患有ASD。在CHR15Q11 – Q13区域中表现出不同程度的自闭症表型的许多ASD具有不同程度的自闭症表型。与ASD相关的许多潜在候选基因都存在于该染色体段中。然而,几个临床,体内和体外研究比随机和无偏的其他研究更频繁地选择一个基因。该基因代码为UBE3A或泛素蛋白连接酶E3A [也称为E6AP泛素蛋白 - 蛋白连接酶(E6AP)],这是一种参与蛋白质细胞降解的酶。该基因已被列为几个基因之一,其在自闭症数据库中引起ASD的潜力很高。UBE3A基因中功能突变,三重或重复的增益也与ASDS诸如Angelman综合征(AS)和DUP15Q综合征等ASD有关。大脑中UBE3A的遗传印迹和对神经母体特异性表达的偏爱是各种ASD的关键特征。由于UBE3A基因参与了与自闭症样症状相关的两种主要重要疾病,因此在理解该基因与自闭症之间的联系时进行了广泛的研究。此外,由于没有通用方法或机制来识别ube3a介导的ASD,因此对于神经生物学家,神经科学家和临床医生设计疗法或诊断工具的神经生物学家,神经科学家和临床医生仍然具有挑战性。在这篇综述中,我们关注UBE3A蛋白的结构和功能方面,讨论ASD的15q11 – Q13区域的主要相关性,并突出显示UBE3A和ASD之间的联系。我们试图通过详细介绍UBE3A介导的ASD的可能机制来扩大读者的知识,从而强调了UBE3A作为ASD的急诊诊断中的前瞻性生物标志物的用法,并讨论了积极的成果,高级发展,先进的发展,高级发展,以及针对UBEBEBEBEBEBEBEBEBEBEBEBEBEBBE3A-ASDS的障碍。本评论是新颖的,因为它为与显示自闭症症状的疾病相关的最重要基因之一提供了一个非常详细且全面的平台。此外,这篇综述还试图对在接下来的几年中对这些UBE3A介导的ASD的诊断,预防和治疗的可能步骤进行乐观反馈。

ASHG 2023 全体会议摘要
使用 CRISPR/Cas9 进行神经遗传疾病的基因编辑面临难以穿过血脑屏障、渗透性有限和治疗窗口狭窄的挑战。虽然改良的腺相关病毒 (AAV) 克服了其中一些障碍,但由于 Cas9 蛋白的长期存在,它们的免疫原性和更高的脱靶效应风险限制了它们对人类神经遗传疾病的转化价值。为了解决这个问题,我们开发了一种创新的非病毒递送工具,使用与 Cas9 蛋白和 sgRNA 结合的化学修饰核糖核蛋白 (RNP) (cRNP-Cas9/sgRNA,cRNPcg)。由于其尺寸小 (12um),cRNPcg 能够有效渗透到大脑中的神经元细胞中,而瞬时 Cas9 蛋白大大降低了脱靶效应的风险。我们在体外和体内测试了 cRNPcg 对 Angelman 综合征 (AS) 的疗效,这是一种由神经元和母体特异性 UBE3A 基因表达缺陷引起的神经发育障碍。父系染色体中 UBE3A 的抑制表达由父系表达的非编码 UBE3A 反义转录本 (UBE3A-ATS) 介导。通过反义寡核苷酸 (ASO) 灭活 UBE3A-ATS 在正在进行的 1/2 期临床试验中显示出积极的临床效果。然而,ASO 的短暂作用需要每月鞘内注射,这对作为标准临床治疗方法提出了挑战。我们设计的 cRNPcg 系统可选择性地灭活 Ube3a-ATS 表达,并可能通过单次治疗实现永久性治疗效果。使用 Ube3a-YFP 报告小鼠,我们观察到高基因编辑效率(>75% 靶向细胞)和广泛的脑渗透。我们给新生儿 (P1-2) 和 P21 AS Ube3 a m-/p+ 模型鞘内注射了 cRNPcg,观察到 Ube3a-ATS 显著降低,并且 Ube3a 重新激活至正常水平的 30%,遍及皮质、海马和小脑。因此,这种治疗显著改善了多个行为领域,包括运动功能、焦虑样行为、学习和记忆,并且还延长了成年 AS Ube3 a m-/p+ 小鼠化学诱发的肌阵挛和强直性癫痫发作的潜伏期。重要的是,我们没有观察到与 cRNPcg 相关的任何急性或慢性毒性。此外,我们发现 cRNPcg 有效地重新激活了 AS 患者 hIPSC 衍生的神经祖细胞中父系染色体上的 UBE3A 表达,这些神经祖细胞存在 15q11-q13 的大量母系缺失。总之,我们的结果表明,cRNPcg 是一个将 CRISPR/Cas9 基因编辑传递到大脑的创新平台,具有广泛的应用和治疗许多其他神经遗传疾病的潜力。
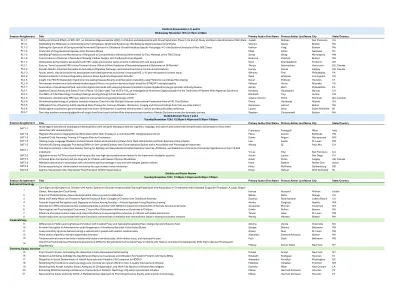
会议任务标题 主要作者名字 主要作者姓氏 城市 州/国家 PL1-1 STK-001 的安全性和临床效果,抗
会议任务标题 主要作者名字 主要作者姓氏 城市 州/国家 PL1-1 STK-001(一种反义寡核苷酸 (ASO))对患有 Dravet 综合征的儿童和青少年的安全性和临床效果:第 1/2a 期研究结束和开放标签扩展 (OLE) 数据 Joseph Sullivan 旧金山 CA Pl1-2 描述癫痫 - 运动障碍综合征的分子和临床谱(癫痫 - 运动障碍谱研究) Vicente Quiroz 波士顿 MA PL1-3 定义儿童期遗传性痉挛性截瘫的锥体外系运动障碍谱:超过 500 例病例的横断面分析 Kathryn Yang 波士顿 MA PL1-4 围产期中风后耐药性癫痫的预测因素 Miles Fisher 纳什维尔 TN PL1-5确定对抽搐综合行为干预的反应预测因子和机制:TReC 研究方法 Sonya Wang 明尼阿波利斯 MN PL1-6 心肺旁路心脏手术后新生儿癫痫发作发生率低 Eleonore Valencia 波士顿 MA PL1-7 青少年和年轻人睡眠期间的 Delta 功率与 MRI 可见的血管周围容积有关 Seva Khambadkone 波特兰 OR PL2-1 极早产儿早期与足月等效 MRI:哪种方法更能预测 36 个月时的神经发育结果? Thiviya Selvanathan 加拿大不列颠哥伦比亚省温哥华 PL2-2 硫酸角质素:人类胎儿前脑神经母细胞迁移途径和轴突束的化学模板 Harvey Sarnat 加拿大阿尔伯塔省卡尔加里 PL2-3 新生儿 HIE 发育结果与种族、民族和社会经济关系:一项为期 10 年的回顾性队列研究。 Whitney Fitts 费城 PA PL2-4 唐氏综合症退化症中的免疫调节基因新生变异 Saba Jafarpour 洛杉矶 CA PL2-5 深入了解 MECP2 重复综合症:使用多组学和深度表型分析揭示疾病严重程度和表达变异性 Davut Pehlivan 休斯顿 TX PL2-6 SYNGAP1 脑病患者体外细胞模型的意外和新型线粒体表型 Melissa Greco 罗阿诺克 VA PL2-7 脑脊液限制性寡克隆带与儿童髓鞘少突胶质细胞糖蛋白抗体病复发性疾病的关联 Ashley Bach 费城 PA PL3-1 GTX-102(一种用于治疗的在研反义寡核苷酸)的 1/2 期开放性试验的最新临床活性和安全性患有 Angelman 综合征的患者 Kimberly Goodspeed Novato CA PL3-2 儿童神经病学 ABC:通过基于文章的课程创造终身学习 Elizabeth Troy Aurora CO PL3-3 偏瘫性脑瘫 MRI 损伤模式和按出生胎龄划分的症状 Johanie Victoria Piché Montreal QC,加拿大 PL3-4 儿童多发性硬化症的全球流行病学:数据来自多发性硬化症国际联盟 MS 图集,第三版 Grace Gombolay 亚特兰大 GA PL3-5 CANaspire 全身 AAV9 介导的 Canavan 病基因治疗试验:低剂量组的生物标志物、影像学和临床发现 Genevieve Laforet 博尔顿 MA PL3-6 Eladocagene exuparvovec 基因疗法改善芳香族 L-氨基酸脱羧酶缺乏症患者的运动发育 Lauren Warn 南普莱恩菲尔德 NJ PL3-7 单干血斑测试的两步新生儿筛查算法识别出不成比例的女性杜氏肌营养不良症携带者 Stephen Chrzanowski 波士顿 MA