XiaoMi-AI文件搜索系统
World File Search SystemG12C
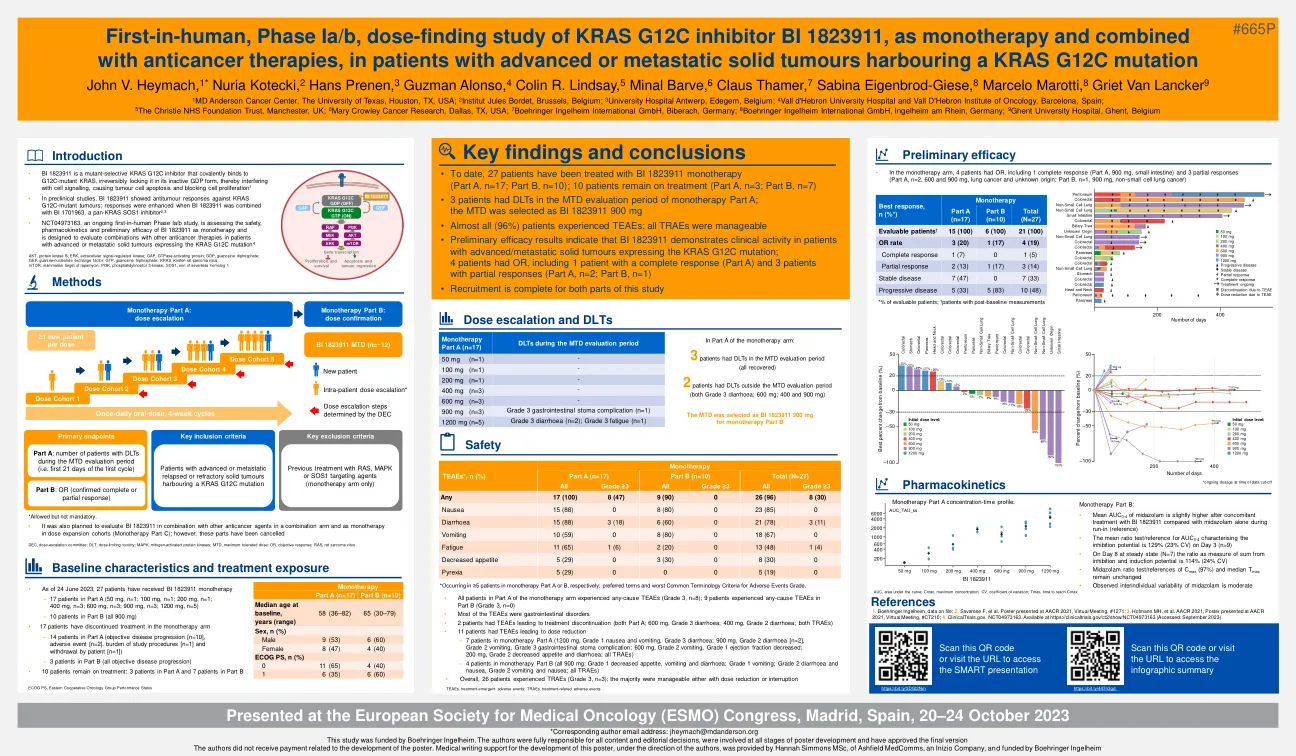
DLL3/CD3 IgG样T细胞Engager obrixtamig*(BI 764532)的I期试验对DLL3阳性肿瘤患者:LCNEC-L
1 MD Anderson癌症中心,德克萨斯大学,美国德克萨斯州休斯敦; 2比利时布鲁塞尔的Jules Bordet Institut; 3比利时Edegem的Antwerp大学医院; 4 Vall D'Hebron大学医院和西班牙巴塞罗那市肿瘤学研究所; 5英国曼彻斯特的Christie NHS基金会信托基金; 6美国德克萨斯州达拉斯的玛丽·克劳利癌症研究; 7 Boehringer Ingelheim International GmbH,德国比伯拉赫; 8 Boehringer Ingelheim International GmbH,Ingelheim Am Rhein,德国; 9根特大学医院,根特,比利时1 MD Anderson癌症中心,德克萨斯大学,美国德克萨斯州休斯敦; 2比利时布鲁塞尔的Jules Bordet Institut; 3比利时Edegem的Antwerp大学医院; 4 Vall D'Hebron大学医院和西班牙巴塞罗那市肿瘤学研究所; 5英国曼彻斯特的Christie NHS基金会信托基金; 6美国德克萨斯州达拉斯的玛丽·克劳利癌症研究; 7 Boehringer Ingelheim International GmbH,德国比伯拉赫; 8 Boehringer Ingelheim International GmbH,Ingelheim Am Rhein,德国; 9根特大学医院,根特,比利时

如何治疗KRAS G12C突变的晚期非小细胞肺癌...
在包括 NSCLC 在内的所有肿瘤类型中,约 98% 的致癌 RAS 突变发生在 Switch I 的 G12 或 G13 密码子上,或 Switch II 区域的 Q61 密码子上。24 这些突变的获得导致 KRAS 活性改变,从而维持不受控制的 KRAS 信号网络并促进肿瘤形成和进展(图 1A、B)。KRAS 中的 G12 突变是最常见的突变,占肺癌中所有 KRAS 突变的近 90%,其次是密码子 13 和 61 的突变。24 新兴证据表明,不同的 KRAS 异构体在临床特征、并发基因组变异和基因表达谱方面高度异质,凸显了不同 KRAS 突变体潜在的异构体依赖性治疗脆弱性。 16 KRAS G12C 突变与烟草暴露密切相关,据报道,与其他 KRAS 亚型和 KRAS 野生型 NSCLC 相比,KRAS G12C 突变具有更高的肿瘤突变负担和较高的基因同时突变率,例如 STK11 、 KEAP1 、 SMARCA4 和 ATM 。16,17 此外,具有 KRAS G12C 突变的 NSCLC 倾向于上调免疫逃逸标志物,例如 PD-L1 和 PD-L2,从而部分解释了在该患者群体中观察到的对 ICI 的敏感性增加。16,25
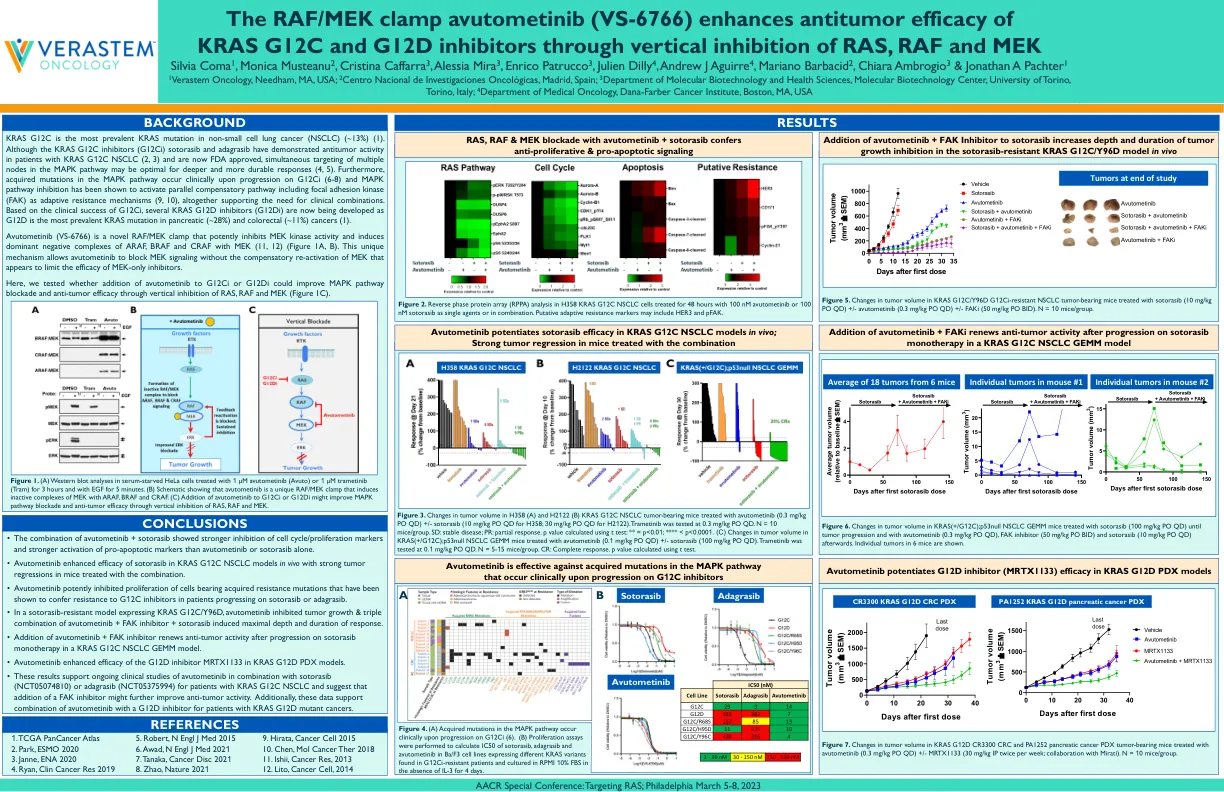
RAF/MEK 钳制 avutometinib (VS-6766) 通过垂直抑制 RAS、RAF 和 MEK 增强 KRAS G12C 和 G12D 抑制剂的抗肿瘤功效
KRAS G12C 是非小细胞肺癌 (NSCLC) 中最常见的 KRAS 突变 (约 13%) (1)。尽管 KRAS G12C 抑制剂 (G12Ci) sotorasib 和 adagrasib 已证明对 KRAS G 1 2C NSCLC 患者具有抗肿瘤活性 (2, 3) 并且现已获得 FDA 批准,但同时靶向 MAPK 通路中的多个节点可能对更深层次和更持久的反应最有利 (4, 5)。此外,MAPK 通路中的获得性突变在 G12Ci 进展时在临床上发生 (6-8),并且 MAPK 通路抑制已被证明可以激活平行补偿通路,包括粘着斑激酶 (FAK) 作为适应性耐药机制 (9, 10),共同支持临床联合用药的必要性。基于 G12Ci 的临床成功,目前正在开发几种 KRAS G12D 抑制剂 (G12Di),因为 G12D 是胰腺癌 (~ 28%) 和结直肠癌 (~ 11%) 中最常见的 KRAS 突变 (1)。

CHD相关增强剂塑造人类心肌细胞谱系承诺
提高对KRAS G12C靶向疗法的抗肿瘤反应的抽象努力从利用组合方法中受益。在这里,我们将SOS1-KRAS相互作用抑制剂BI-3406诱导的抗肿瘤反应与KRAS G12C抑制剂(KRAS G12C I)与KRAS G12C i单独或与SHP2或EGFR抑制剂合并的抗肿瘤反应。在肺癌和结直肠癌(CRC)模型中,BI-3406加上KRAS G12C I诱导抗肿瘤反应比单独使用KRAS G12C I观察到的抗肿瘤反应更强,并且与其他组合相比。这种增强的抗肿瘤反应与RAS-MAPK信号的更强,更扩展的抑制作用有关。重要的是,BI-3406加KRAS G12C I治疗延迟了CRC和肺癌模型中获得的Adagrasib耐药性的出现,并且与KRAS G12C I-抗性CRC模型中抗增殖活性的重新建立有关。我们的发现位置KRAS G12C加SOS1抑制疗法是治疗KRAS G12C肿瘤的有前途的策略,以及解决对KRAS G12C I的获得性抗性。

癌症
开发出新的替代疗法。多中心临床试验的令人满意的结果促使 KRAS G12C 抑制剂疗法最近获得批准。尽管 KRAS G12C 等位基因特异性药物极大地改善了 KRAS G12C 肿瘤患者的临床前景,特别是肺腺癌患者,其中 KRAS G12C 突变体与其他 KRAS 突变相比最为普遍,但必须克服不可避免的挑战,例如内在和获得性耐药性,以最大限度地发挥 KRAS G12C 抑制剂疗法的功效。最近的研究表明,补偿性信号通路(例如 PI3K/AKT/mTOR 通路)和表观遗传重编程(例如上皮间质转化 (EMT))是介导对 KRAS G12C 抑制剂的内在耐药性的常见机制,而当癌细胞获得 KRAS 蛋白的二次突变,从而削弱 KRAS G12C 抑制剂的共价结合时,可能会产生获得性耐药性和随之而来的复发性疾病。识别和靶向 KRAS G12C 抑制剂耐药机制有望为有效治疗 KRAS G12C 突变型癌症患者提供新策略。

针对KRASG12C的突破
开发出新的替代疗法。多中心临床试验的令人满意的结果促使 KRAS G12C 抑制剂疗法最近获得批准。尽管 KRAS G12C 等位基因特异性药物极大地改善了 KRAS G12C 肿瘤患者的临床前景,特别是肺腺癌患者,其中 KRAS G12C 突变体与其他 KRAS 突变相比最为普遍,但必须克服不可避免的挑战,例如内在和获得性耐药性,以最大限度地发挥 KRAS G12C 抑制剂疗法的功效。最近的研究表明,补偿性信号通路(例如 PI3K/AKT/mTOR 通路)和表观遗传重编程(例如上皮间质转化 (EMT))是介导对 KRAS G12C 抑制剂的内在耐药性的常见机制,而当癌细胞获得 KRAS 蛋白的二次突变,从而削弱 KRAS G12C 抑制剂的共价结合时,可能会产生获得性耐药性和随之而来的复发性疾病。识别和靶向 KRAS G12C 抑制剂耐药机制有望为有效治疗 KRAS G12C 突变型癌症患者提供新策略。

抑制ULK1/2和KRASG12C控制肿瘤生长... 供体衍生的多重白血病抗原特异性T细胞疗法,以防止所有患者移植后复发 与年龄相关的丁酸酯产生细菌的时间下降在3×TG-AD小鼠的神经病理学和记忆缺陷的发作和进展中起关键的致病作用 naltrexone-bupropion组合不影响可卡因自我 胰腺内导管内乳头肿瘤的空间转录组将NKX6-2鉴定为胃分化和顽固生物潜能的驱动力 昼夜节律作为发育期间和整个衰老期间大脑健康的调节剂 小鼠心脏发育的三维微观成像从早期植入后到晚期阶段 使用现场虚拟培训课程改善学校卫生工作者的糖尿病管理
KRAS的摘要突变激活通常发生在肺癌发生中,并且随着美国食品和药物管理局最近批准KRAS G12C的共价抑制剂,例如Sotorasib或Adagrasib,KRAS癌蛋白是非小细胞肺癌(NSCLC)的重要药理靶标。但是,并非所有KRAS G12C驱动的NSCLC都对这些抑制剂做出反应,并且那些反应反应的患者的耐药性出现可能是迅速而多效的。因此,基于共价抑制KRAS G12C的支柱,正在努力开发有效的组合疗法。在这里,我们报告说,KRAS G12C信号传导的抑制会增加KRAS G12C表达肺癌细胞的自噬。此外,DCC -3116(一种选择性ULK1/2抑制剂)的组合以及sotorasib显示了对人Kras G12C驱动的肺癌细胞增殖的合作/协同抑制体内体外和肿瘤对照中的抑制作用。此外,在KRAS G12C驱动的NSCLC的基因工程小鼠模型中,抑制KRAS G12C或ULK1/2的抑制会减轻肿瘤负担并增加小鼠的存活率。因此,这些数据表明ULK1/2介导的自噬是对肺癌中KRAS G12C抑制的药理作用的细胞保护胁迫反应。
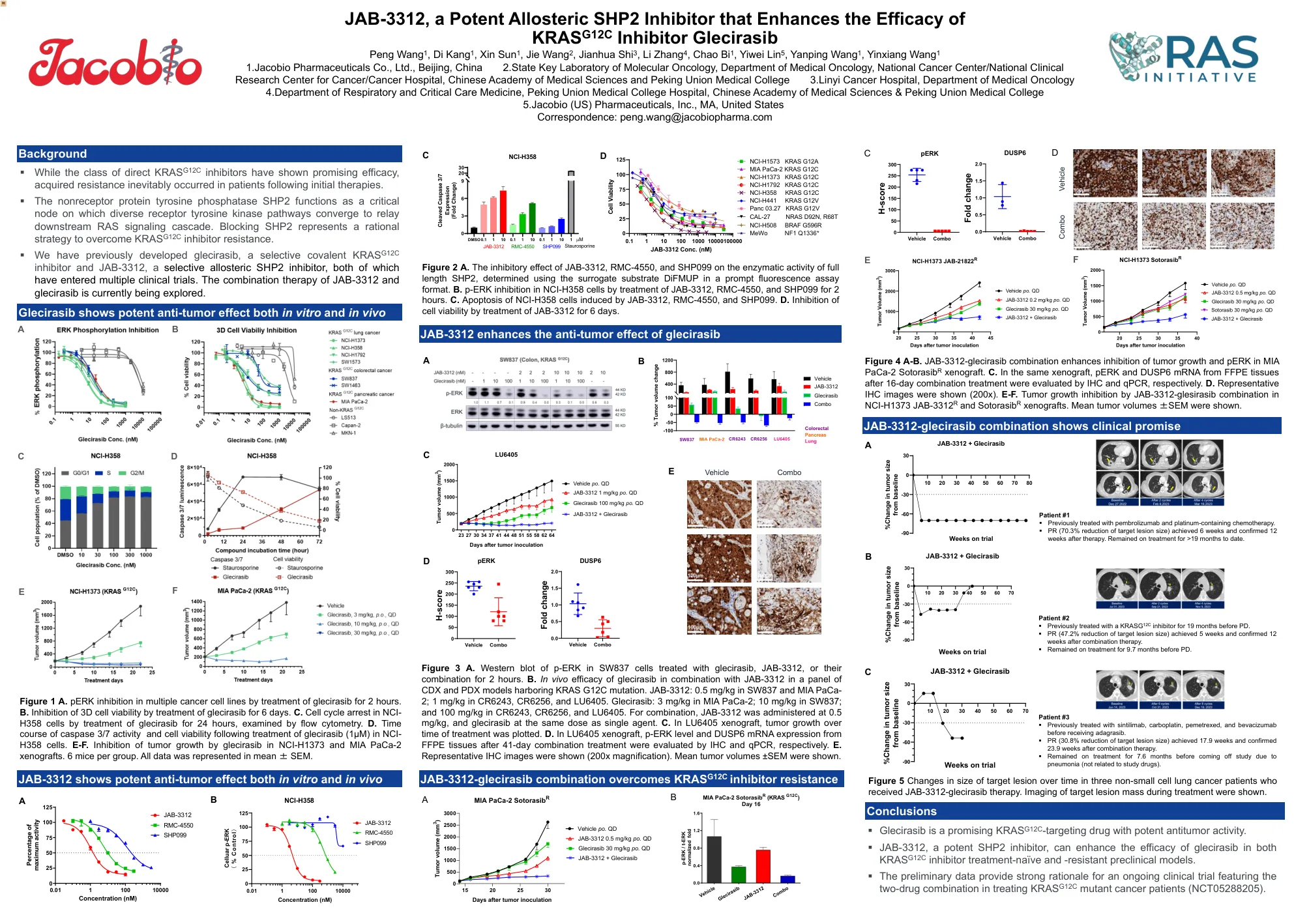
JAB-3312,一种有效的变构 SHP2 抑制剂,可增强 KRAS G12C 抑制剂 Glecirasib 的疗效 王鹏 1 ,康迪 1 ,孙鑫 1 ,魏杰
图 3 A. 用格列西拉西、JAB-3312 或二者联合处理 SW837 细胞 2 小时后,p-ERK 的蛋白质印迹分析。B. 格列西拉西与 JAB-3312 联合用于携带 KRAS G12C 突变的一组 CDX 和 PDX 模型的体内疗效。JAB-3312:SW837 和 MIA PaCa-2 中为 0.5 mg/kg;CR6243、CR6256 和 LU6405 中为 1 mg/kg。格列西拉西:MIA PaCa-2 中为 3 mg/kg;SW837 中为 10 mg/kg;CR6243、CR6256 和 LU6405 中为 100 mg/kg。联合用药中,JAB-3312 剂量为 0.5 mg/kg,格列西拉西与单药剂量相同。 C. 在 LU6405 异种移植中,绘制了肿瘤生长随治疗时间的变化。D. 在 LU6405 异种移植中,分别通过 IHC 和 qPCR 评估 41 天联合治疗后 FFPE 组织的 p-ERK 水平和 DUSP6 mRNA 表达。E. 显示了代表性 IHC 图像(放大 200 倍)。显示了平均肿瘤体积 ±SEM。

KRASG12C抑制剂JAB-21822与...
(A-B)示意图,表明RTK/SHP2介导的MAPK途径重新激活是KRAS G12C抑制剂耐药性的关键机制。将SHP2抑制剂与KRAS G12C抑制剂铅组合在MAPK途径活性的最大下调(C)KRAS G12C抑制剂R MIA PACA-2细胞系中,通过JAB-21822和JAB-3312的组合在不同的浓度下,用JAB-21822和JAB-3312组合评估了SYSS SYSIS抑制作用(JAB-21822和JAB-3312)的抑制作用(D)。 KRAS G12C抑制剂R NCI-H358细胞系(E)的JAB-3312组合(E)log 2折叠NCI-H358细胞中基因表达的变化,具有对KRAS G12C抑制剂的耐药性,通过RNaseq(F)获得了KRAS G12C抑制剂的耐药性,而NCI-H358细胞中的NCI-H358细胞中的基因表达水平

通过新型 KRAS Switch-II 口袋突变和多克隆变异(以 RAS–MAPK 再激活为中心)获得对 KRASG12C 抑制的临床获得性耐药性
摘要 突变选择性 KRAS G12C 抑制剂,例如 MRTX849 (adagrasib) 和 AMG 510 (sotorasib),已证明对 KRAS G12C 突变癌症(包括非小细胞肺癌 (NSCLC))有效。然而,临床获得性耐药 KRAS G12C 抑制剂的潜在机制仍未确定。为了开始定义获得性耐药的机制谱,我们描述了一名患有 KRAS G12C NSCLC 的患者,该患者对 MRTX849 产生了多克隆获得性耐药,在四个基因(KRAS、NRAS、BRAF、MAP2K1)的连续无细胞 DNA 中出现了 10 种异质性耐药性改变,所有这些改变都汇聚在一起重新激活 RAS-MAPK 信号传导。值得注意的是,研究人员发现一种新的 KRAS Y96D 突变会影响 MRTX849 和其他非活性状态抑制剂结合的 switch-II 口袋,这种突变会干扰关键的蛋白质-药物相互作用,并在工程化和患者衍生的 KRAS G12C 癌症模型中产生对这些抑制剂的耐药性。有趣的是,一种功能独特的新型三重复合物 KRAS G12C 活性状态抑制剂 RM-018 保留了结合和抑制 KRAS G12C/Y96D 的能力,并且可以克服耐药性。