XiaoMi-AI文件搜索系统
World File Search SystemSBLA

Dupixent® sBLA 获 FDA 优先审查...
∗ 基于两项 3 期试验的积极结果,获得优先审查 ∗ 如果获得批准,Dupixent 将成为 COPD 的唯一生物疗法,也是十多年来该疾病的首个新治疗方法 ∗ 中国和欧洲的监管提交也在接受审查 巴黎和纽约州塔里敦 2024 年 2 月 23 日。美国食品药品监督管理局 (FDA) 已接受优先审查 Dupixent ® (dupilumab) 的补充生物制品许可申请 (sBLA),作为某些未受控制的慢性阻塞性肺病 (COPD) 成年患者的附加维持治疗,这是第六个潜在适应症。FDA 决定的目标行动日期为 2024 年 6 月 27 日。中国和欧盟的监管提交也在接受审查。sBLA 以及世界各地的其他提交申请均由 3 期 COPD 临床研究计划的数据支持,该计划评估了 Dupixent 对目前或以前吸烟且患有未控制的 COPD 且有 2 型炎症证据的成年人的疗效和安全性(筛查血液嗜酸性粒细胞 >300 个细胞/微升)。所有患者均接受背景最大限度标准护理吸入疗法(几乎所有患者均接受三联疗法)。两项试验(BOREAS、NOTUS)均达到了主要终点,表明与安慰剂相比,Dupixent 分别显着降低了年中度或重度急性 COPD 发作率 30% 和 34%。在两项试验中,与安慰剂相比,Dupixent 还迅速显着改善了肺功能,并且改善持续到 52 周。两项试验的安全性结果与 Dupixent 在其获批适应症中的已知安全性基本一致。在两项试验中,与安慰剂相比,Dupixent 更常见的不良事件(≥5%)是背痛、COVID-19、腹泻、头痛和鼻咽炎。优先审查授予寻求批准的监管申请,这些申请有可能在治疗、诊断或预防严重疾病方面提供显着改善。Dupixent 在 COPD 中的潜在用途目前正在临床开发中,其针对该适应症的安全性和有效性尚未得到任何监管机构的充分评估。关于 COPD COPD 是一种呼吸系统疾病,会损害肺部并导致肺功能逐渐下降。症状包括持续咳嗽、呼吸困难和过多粘液分泌,这不仅可能损害日常活动的能力,还可能导致焦虑、抑郁和睡眠障碍。COPD 还会给健康和经济带来沉重负担,因为反复急性发作需要全身性皮质类固醇治疗和/或导致住院治疗。吸烟和接触有害颗粒是 COPD 的主要危险因素,但即使是戒烟者也可能患上或继续患病。目前还没有新的治疗方法

新闻稿dupixent SBLA接受了FDA优先审查的靶向治疗,以对大胆的pemphigoid
•如果获得批准,dupixent将是美国第一个也是唯一的有针对性药物治疗BP的药物; FDA decision expected by June 20, 2025 • Priority review granted based on positive pivotal results demonstrating significant improvements in sustained disease remission with Dupixent compared to placebo • BP is a chronic, debilitating and relapsing skin disease with underlying type 2 inflammation characterized by intense itch and blisters, reddening of the skin and painful lesions Paris and Tarrytown, NY, February 18, 2025.美国食品药品监督管理局(FDA)已接受优先审查Dupixent(Dupilumab)的补充生物制品申请(SBLA),以用大胆的Pemphigoid(BP)治疗成年人。SBLA受到一项关键研究的数据支持,该研究评估了106名中度至重度BP成年人的Dupixent的功效和安全性。与安慰剂相比,达到了主要终点,五倍的夫人患者可以增加持续的疾病缓解。持续的疾病缓解被定义为完全临床缓解,通过在第16周完成口服皮质类固醇(OCS)锥度(OCS)锥度(OCS OCT治疗,仅在36周治疗期间不复发且无需救援治疗)而无需复发治疗。这项研究还表明,与安慰剂相比,偶像可显着降低疾病的严重程度,瘙痒和OCS的使用。不良事件更常见(至少3例患者)包括外周水肿,腹痛,背痛,视力模糊,高血压,哮喘,结膜炎,便秘,上呼吸道,上呼吸道感染,肢体感染,LIMB损伤和失眠症。关于dupixentbp是一种慢性,令人衰弱和复发性皮肤病,其潜在的2型炎症通常发生在老年人群中。它的特征是强烈的瘙痒和水泡,皮肤发红以及疼痛的病变。水泡和皮疹会在大部分身体上形成,并导致皮肤出血和外壳,从而导致患者更容易感染并影响其日常功能。美国大约有27,000名成年人生活在全身性皮质类固醇中无法控制的BP。优先审查授予寻求批准疗法的监管应用,这些疗法有可能在治疗,诊断或预防严重状况方面有显着改善。dupixent以前曾被FDA授予BP的孤儿药物名称,该药物适用于用于治疗美国少于20万人的稀有疾病的研究药物。dupixent在BP中的安全性和功效目前正在临床评估中,尚未由任何监管机构评估。

蜂窝,组织和基因疗法咨询委员会2024年11月21日会议草案
委员会将在公开会议上开会,讨论并提出有关Astra Zeneca的补充生物制品申请(SBLA)125586/546,以确认Andexxa的临床益处(cogaulational XA(重组),与Revaroxaban或Aperagn的患者相关的患者,co凝结因子XA(重组),灭活-ZHZO)的临床益处。不受控制的出血。

leqembi™(lecanemab-imb) - FDA接受...
- 根据第2阶段数据的加速批准,Leqembi获得了批准,这表明Leqembi减少了淀粉样蛋白β斑块在大脑中的积累。- 在验证性试验中,持续批准取决于Leqembi对Leqembi的临床益处的验证。•将加速批准转换为传统批准的SBLA基于第3阶段的发现,即Clarity AD试验。在该研究中,Leqembi通过减少临床痴呆率评级 - 盒子(CDR -SB)评分(CDR -SB)得分的临床下降来达到主要终点,而安慰剂则代表治疗差-0.45(p <0.001)。

首份 BrainTransporter 技术协议签署 - BioArctic
第一季度末之后的事件 • BioArctic 的合作伙伴 Eisai 向美国食品药品监督管理局 (FDA) 提交了一份补充生物制品许可申请 (sBLA),用于使用 lecanemab 进行较低频率的静脉 (IV) 维持给药 • BioArctic 被纳入纳斯达克斯德哥尔摩的新 ESG 责任指数 • BioArctic 和 Eisai 就 BAN2802 达成了一项研究评估协议,BAN2802 是一种潜在的新疗法,将 BioArctic 专有的 BrainTransporter™ 技术与阿尔茨海默氏症候选药物相结合 • Eisai 获得快速通道资格并向 FDA 启动了滚动生物制品许可申请 (BLA),用于 Leqembi 的皮下维持给药 • Eisai 公布了 Leqembi 2024 财年(2024 年 4 月 - 2025 年 3 月)的销售预测为 565 亿日元

统计审查-Everidys
AAV Adeno-Associated Virus AE Adverse Event AESI Adverse Event of Special Interest BLA Biologics License Application CI Confidence Interval DMD Duchenne Muscular Dystrophy FDA Food and Drug Administration IR Information Request ISE Integrated Summary of Efficacy ISS Integrated Summary of Safety ITT Intent-to-Treat IV Intra-Venous LSM Least Squares Mean MAR Missing At Random mITT Modified Intent-to-Treat MMRM Mixed Model for Repeated Measures MNAR Missing Not At Random MWR Meter Walk/Run NSAA North Star Ambulatory Assessment PROMIS Patient-Reported Outcomes Measurement Information System REML Restricted Maximum Likelihood SAE Serious Adverse Event SAP Statistical Analysis Plan SAS Statistical Analysis System sBLA Supplemental Biologics License Application SE Standard Error SV95C Stride Velocity 95th Centile TEAE Treatment-Emergent Adverse Event

2024年5月7日临床审查备忘录-Altuviiio
在批准时,先前治疗的小儿患者(EFC16295; NCT04759131)进行了关键的第三阶段研究,并进行了临时疗效和安全数据以进行审查。提交补充生物制剂申请(SBLA)提供了针对EFC16295研究的最终临床研究报告,以及第三阶段研究的合并手术和安全数据。由于BIVV001当前已被批准为儿科受试者,因此该提交为小儿研究提供了更新的功效和安全性数据,该研究已纳入标签中。完成的研究EFC16295是根据SBLA提交评估的主要研究。研究EFC16295是一项多中心,开放标签的研究,用于评估BIVV001治疗的PK,安全性和疗效,用于预防和治疗先前治疗的小儿受试者(<12岁)的出血。在儿科研究中总共有74名先前治疗的受试者中评估了BIVV001的安全性和功效,他们接受了至少一剂BIVV001。这包括另外51名进行分析的受试者,因为临时分析仅包括具有至少26周(n = 23 = 23周)的受试者。有74名受BIVV001治疗的受试者<12岁。七十三个治疗的疗效期限超过26周。七十二名受试者是可以评估的功效,并具有平均年化出血率(ABR)(95%CI)为2.6(1.6,4.0),ABR的中位数为0.5(0,2.1),为所有出血。最常见的AE是温和且短暂的上呼吸道感染和上级。未检测到抑制剂。与BIVV001给药前基线平均处理的ABR(标准偏差; SD)为2.1(4.2)和中位数为1.0(0; 32)相比,所处理的ABR为0.6(95%CI:0.4、0.9)和ABR(Q1,Q3)的中位数为0(0,1.0),为0(0,1.0)。小儿人群中没有发现新的安全信号。评估了两名需要两种主要手术程序的受试者的围手术期管理,并用BIVV001治疗了手术止血。用BIVV001治疗在两项主要手术中都提供了良好或出色的止血控制。由于孤儿的名称,此提交并未触发小儿研究权益法(PREA)。没有售后承诺或要求。

统计评论 STN: 125742/276 第 i 页
BioNTech 和申请人辉瑞提交了一份补充生物制品许可申请 (sBLA),STN 125742/276,用于 BNT162b2 疫苗的主动免疫,以预防 12 岁及以上人群中由严重急性呼吸道综合征冠状病毒 2 (SARS-CoV-2) 引起的 2019 年冠状病毒病 (COVID-19)。最初的提交是为了寻求批准 30 μg 剂量水平的 BNT162b2 二价 (Original 和 Omicron BA.4/BA.5) 疫苗作为 2 剂初级系列和加强剂。提交的材料包括免疫原性数据(即针对 Omicron BA.4/BA.5 和参考菌株的中和滴度)和安全性数据,这些数据来自研究 C4591044 的第 2 组(≥12 岁)和第 3 组(≥18 岁)参与者的安全性数据,这些参与者在接种 3 剂 BNT162b2 后,以 30 或 60 μg 的剂量作为加强剂接种 BNT162b2 二价疫苗 (WT/OMI BA.4/BA.5)。2023 年 4 月 18 日,美国食品药品监督管理局 (FDA) 修改了辉瑞-BioNTech COVID-19 二价疫苗的紧急使用授权 (EUA),以将疫苗接种计划简化为对大多数人使用单剂,此后,此 sBLA 的意图被修改为寻求授权单剂使用,无论之前的疫苗接种状况如何。 2023 年 6 月 2 日,CBER 在 CBER 与申请人的电话会议中请求对 12 岁及以上的 COVID-19 疫苗初治、基线 SARS-CoV-2 阳性个体进行单剂量研究。2023 年 6 月 16 日,FDA 建议辉瑞/BioNTech 根据 2023 年 6 月 15 日举行的疫苗和相关生物制品 (VRBPAC) 会议关于 2023-2024 年 COVID-19 疫苗配方的 SARS-CoV-2 毒株组成,开发一种单价(XBB.1.5 变体)COVID-19 疫苗,供潜在合格人群使用。 2023 年 6 月 23 日,申请人向 STN 125742/276.16 提交了更新后的方案,以支持 30 μg COMIRNATY 2023-2024 配方的许可,供年龄 ≥12 岁的个人使用,无论之前的 COVID-19 疫苗接种情况如何。申请人向 STN 125742/276.25 提交了来自 BNT162-17 研究的数据,以支持在未接种过 COVID-19 疫苗的血清阳性个体中使用单剂量 COMIRNATY,其中包括在年龄≥18 岁至≤85 岁且有先前感染 SARS-CoV-2 证据的未接种过 COVID19 疫苗的参与者中单剂量 30μg 改良二价 BNT162b2(B.1.1.7 + B.1.617.2 或 Alpha/Delta 株)疫苗后的免疫原性结果,以及在研究 C4591001 中接受 2 剂 30 μg 原始 BNT162b2 疫苗且没有 SARS-CoV-2 感染证据的 COVID19 疫苗初次参与者的比较子集的免疫原性结果。作为批准后的承诺,申请人提议对 C4591054 进行额外的子研究(子研究 B),其中先前暴露了 SARS-CoV-2,12 岁及以上的未接种过疫苗的个人将在接种 30 μg 单价(XBB.1.5 变体)COVID-19 疫苗 1 个月后接受免疫原性评估。

说话者传记
Jeffrey Siegel,医学博士|办公室主任|药物评估科学办公室(ODES)|新药办公室(OND)| cder | FDA以前Siegel博士从2019年3月至2020年12月,吉利德科学炎症组临床研究主管执行主任。在此之前,Siegel博士曾是2010年至2019年Genentech/Roche产品开发免疫学的风湿病学和稀有疾病全球疾病的高级医疗总监。在Genentech/Roche时,他的小组启动并完成了第2阶段和第3阶段的托库珠单抗(Actemra)(Actemra)的系统性硬化症,获得了2种突破性治疗名称,并提交了2个SBLA,两者都获得了批准 - 并获得了2个孤儿药。Siegel博士在克利夫兰大学医院获得耶鲁大学医学,实习和住院医师培训的医学学位,克利夫兰大学医院的风湿病学培训以及免疫学和信号转导的NIH基础科学研究培训。奖学金后,他加入了海军医学研究所5年,在那里担任了分支机构,信号转导。然后,他加入了FDA,并在那里工作了14年,担任风湿病学临床团队负责人监督医疗官。
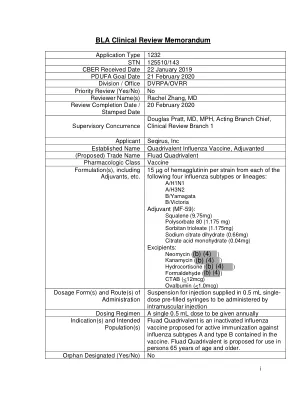
四价氟化物
AE 不良事件 AESI 特别关注的不良事件 aQIV 佐剂四价流感疫苗 AR 不良反应 aTIV 佐剂三价流感疫苗 BIMO CBER 生物研究监测 BLA 生物制品许可证申请 CBER 生物制品评价与研究中心 CFR 联邦法规 CI 置信区间 CMC 化学、制造与控制 CRF 病例报告表 CSR 临床研究报告 FAS 完整分析集 FDA 食品药品管理局 GMT 几何平均滴度 HA 血凝素 HI 血凝抑制 ICH 国际协调会议 ILI 流感样疾病 LL 下限 MedDRA 监管活动医学词典 NOCD 新发慢性病 OBE 生物统计学和流行病学办公室 OVRR 疫苗研究与审查办公室 PeRC 儿科审查委员会 PI 说明书 PMC 上市后承诺 PMR 上市后要求 PPS 按照方案集PREA 儿科研究公平法案 PT 首选术语 QIV 四价流感疫苗 RT-PCR 逆转录聚合酶链反应 SAE 严重不良事件 sBLA 补充生物制品许可申请 SCR 血清转化率 SOC 系统器官分类 STN 提交追踪编号 US 美国 WHO 世界卫生组织