XiaoMi-AI文件搜索系统
World File Search Systemautosomal

Spinraza(Nusinersen)
脊柱肌肉萎缩(SMA)是一种常染色体隐性遗传疾病2,影响10,000例活产的1分之一,并且是婴儿死亡率的最常见遗传原因。大多数病例是由位于5q13处的生存运动神经元1(SMN1)基因中的突变引起的(95%的SMN1外显子7或从SMN1到SMN2的基因转换的纯合缺失)。这些突变会导致运动神经元的进行性变性,从而导致肌肉萎缩。下肢中对刺激的无力和对刺激的反应丧失是最常见的,但是在严重的情况下,控制口腔,喉咙和呼吸的肌肉也可能受到影响。医疗保健专注于呼吸支持,营养支持,抗生素呼吸道感染的管理以及通过支撑,物理治疗和手术的肌腱染色体和脊柱侧弯的管理。尽管最多的寿命


基因编辑剂的治疗性体内递送
精确操纵和编辑人类细胞 DNA 序列的能力可以催生出强大的新型基因组药物。全球有数百万人患有遗传性疾病(Korf 等人,2019 年),其根本原因原则上可以通过治疗性 DNA 编辑剂来纠正。虽然传统的基因增强疗法可以通过提供基因的功能性副本来治疗某些常染色体隐性或单倍体不足性疾病,但基因编辑疗法可以直接纠正基因组 DNA 中的致病突变。因此,原则上,基因编辑可以治疗更广泛的遗传疾病,包括常染色体显性遗传病、因基因产物过少或过多而引起的疾病,或其他简单的基因过度表达无法最佳挽救疾病的疾病。即使对于可以通过现有基因增强或基因沉默策略解决的疾病,通过安装突变来增加或减少靶基因表达的基因编辑疗法也可以通过一次性治疗达到相同的效果,从而提供永久治愈的可能性。更广泛地说,即使没有致病突变的个体,患某些主要疾病(如冠心病)的风险也可以通过精确修改靶基因来调节,这使得基因编辑(如果被证明足够安全有效)有朝一日可能用于降低普通人群的疾病风险。治疗性基因编辑的前景促使人们做出巨大努力将基因编辑疗法引入临床。最近的进展包括开发用于哺乳动物细胞基因编辑的强大工具,包括可编程核酸酶、碱基编辑器和引发编辑器(Anzalone 等人,2020 年;Doudna,2020 年;Newby 和 Liu,2021 年)。这些基因编辑剂具有

VWA2中纯合子和复合杂合的错义突变对阿尔茨海默氏病的贡献
阿尔茨海默氏病是对家族和零星患者的早期(65岁)和晚期(> 65岁)的神经退行性痴呆症的诊断。3种常染色体显性阿尔茨海默氏症基因中的因果突变,即淀粉样蛋白前体蛋白(APP),Presenilin 1(PSEN1)和Presenilin 2(PSEN2),仅解释了5%E 10%E 10%的早期患者,使大多数患者遗传均未解决。为了发现潜在的遗传学缺失,我们使用了17例早期发病患者的全基因组测序数据,该数据有据可查的阿尔茨海默氏病临床诊断。在发现组中,平均发作年龄为55.71 6.83岁(范围37 E 65)。六名患者患有脑尸检和神经疾病,确认了阿尔茨海默氏病。对一名患者识别的遗传数据的分析是von willebrand因子A含有2个基因的域(VWA2)的同伴P.V366M的错义突变(VWA2)。在阿尔茨海默氏病患者队列中对VWA2编码区域的重新判处来自Flanders-Belgium(N¼1148),包括152例早期和996例晚期患者,确定了1个早期和3例晚期患者的额外纯合和复合杂合的杂合杂质性突变。等位基因共享分析在复合杂合VWA2突变载体之间识别出常见的单倍型,这表明共享祖先。总体而言,我们鉴定了5个纯合或复合杂合的错义突变(5/1165; 0.43%)的患者载体,早期(2/169; 1.18%)和3例患者在晚期发作(3/996; 0.30%)患者。2020 Elsevier Inc.保留所有权利。患者中纯合和复合杂合的错义突变的频率高于根据其组合单位等位基因计算得出的频率的预期。纯合/复合杂合的错义突变携带者都没有常染色体显性阿尔茨海默氏病的家族史。我们的发现表明,VWA2中的纯合和复合杂合的错义突变可能有助于偶发患者患阿尔茨海默氏病的风险。

SHORT 综合征引起的非典型糖尿病:一例病例报告
身材矮小、关节过伸、低眼压、Rieger 异常和牙齿萌出延迟 (SHORT) 综合征是一种罕见的原发性常染色体显性遗传病,主要由磷酸肌醇 3-激酶调节亚基 1 (PIK3R1) 基因的致病性功能丧失变异引起。我们报告了一例患有 SHORT 综合征的中国成年女性患者的病例,该患者携带 PIK3R1 基因变异 (c.1945C > T),在 9 年内出现糖代谢异常和严重的餐后胰岛素抵抗。虽然目前尚无针对 SHORT 综合征患者胰岛素抵抗的既定治疗指南,但我们实施了综合治疗计划,包括生活方式干预、二甲双胍和伏格列波糖控制血糖。经过 6 个月的持续观察,患者的血糖水平和胰岛素抵抗显着改善。该案例研究为未来的治疗策略提供了有用的见解。

一名患有冯·希佩尔综合征的 19 个月大儿童出现炎性关节炎......
摘要 冯·希佩尔-林道病 (VHL) 是一种罕见的常染色体显性遗传病,其特征是逐渐形成囊肿和肿瘤。幼年特发性关节炎 (JIA) 是一种慢性炎症性疾病,也是儿童中最常见的关节炎。尽管其发病机制尚不完全清楚,但 JIA 被认为是一种多基因自身免疫介导的疾病。遗传或获得性疾病导致免疫失调,可导致肿瘤和自身免疫性疾病,但文献中很少有报道患有 VHL 和伴随自身免疫性疾病的患者病例。本文,我们据我们所知描述了第一例患有 VHL 和炎症性关节炎的儿童病例,并讨论了可能将 VHL 和 JIA 联系起来的三种可能的病理生理机制。了解这两种疾病共同的病理生理学和遗传学可能有助于指导未来的靶向治疗方向,并改善临床结果。

与患者谈论肥厚性心肌病
•HCM是最常见的遗传性心脏疾病。•通常以常染色体优势模式继承,但不需要HCM的家族史。•许多人不知道自己拥有它或携带遗传易感性。•HCM可以以不同程度的严重程度显示出大不相同的表现,即使在同一家族的成员中也是如此。•某些HCM的情况是非孟德尔式的,可能是由多基因风险等位基因和非遗传因素(例如老年,高血压,代谢综合征和肥胖症)组合而产生的。•人们在休息时可能是非刺激性的,但会因运动而变得阻碍性,因此,作为常规评估的一部分,患者进行运动压力超声心动图很重要。•有各种表型和结构性亚型可以为遗传风险和治疗提供依据。•儿童和成人患者之间的SCD风险分层存在重要差异。

selumetinib selumetinib在神经蛋白酶型I型中的不成比例不良事件信号:Faers的见解
神经纤维瘤1型(NF1)或冯·鲁克林氏病是由NF1基因突变引起的一种神经遗传学疾病,以常染色体显性占主导地位的方式遗传(Ibrahim等,2024; Cichowski等,2003)。它的全球发病率约为3,000名,其中约50%是由家族突变引起的。nf1通常带有咖啡馆café-au-lait macules,多个神经纤维,腋窝或腹股沟雀斑,神经纤维瘤是最常见的症状(Legius等人,2021年)。大约20% - 50%的患者出现神经纤维瘤(PNS),会导致疼痛,神经功能障碍,骨骼畸形和脱离(Colombo等,2022)。尽管手术仍然是主要治疗方法,但由于PN和相邻组织之间的不明确边界,完整的手术切除术是具有挑战性的。此外,

通过菲律宾新生儿筛查计划诊断为囊性纤维化的第一个菲律宾婴儿的病例报告
囊性纤维化(CF)是一种由囊性纤维化跨膜电导调节剂(CFTR)基因的突变引起的常染色体隐性疾病,该基因位于7号染色体上,在亚洲人群中很少见。来自日本,中国,约旦和巴林的报告表明,分别表现出1:350,000、1:153,825、1:2,500和1:5,000的流行率,表明了不同亚洲子组之间的差异很大。1-3遗传学可能是这种可变性的原因,因为即使在美国,不同民族人口的流行率也有所不同,其中,高加索人的患病率在1:3,200中,在西班牙裔中1:10,000,西班牙裔美国人中的1:10,500,在西班牙人中,非裔美国人中的1:15,000,1:30,000中的1:30,000。2虽然菲律宾关于CF的患病率尚不清楚,但以前通过美国的加利福尼亚新生儿筛查计划诊断出了五个菲律宾人。4
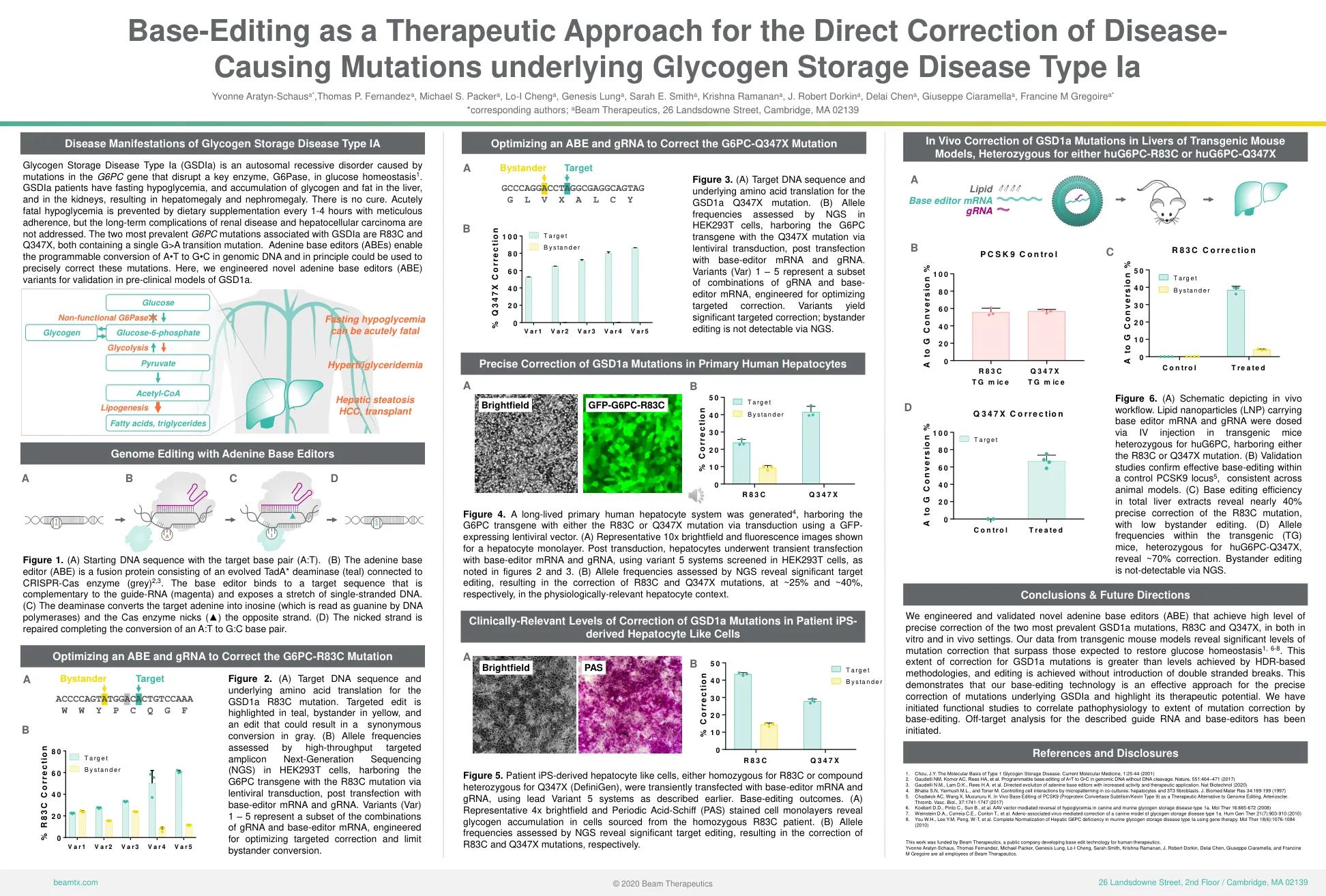
优化ABE和GRNA以纠正G6PC-R83C突变
糖原储存疾病IA型(GSDIA)是由G6PC基因突变引起的常染色体隐性疾病,它破坏了葡萄糖稳态中的关键酶G6Pase 1。GSDIA患者患有低血糖,肝脏和肾脏的糖原和脂肪的积累,导致肝肿大和肾肿大。无法治愈。急性致命的低血糖,但肾脏疾病和肝细胞癌的长期并发症并未解决。与GSDIA相关的两个最普遍的G6PC突变是R83C和Q347X,均包含单个G> A的过渡突变。腺嘌呤碱基编辑器(ABES)可以使用基因组DNA中A•T到G•C的编程转换,并且原理可以用来精确纠正这些突变。在这里,我们设计了新颖的腺嘌呤基础编辑器(ABE)变体,以验证GSD1A的临床模型。