XiaoMi-AI文件搜索系统
World File Search Systemcauses

曲马多会导致体重增加、尼氏物质和星形胶质细胞...
摘要 盐酸曲马多是一种具有中枢作用的合成阿片类药物,用于治疗中度至中度重度疼痛,据报道具有神经毒性。因此,本研究探讨了曲马多对海马结构中体重、尼氏体和星形胶质细胞变化的影响。对照组大鼠口服2ml/kg蒸馏水,第2组大鼠口服50mg/kg曲马多,连续21天。实验前后称量大鼠体重。对大鼠实施安乐死,取脑并称重。将取下的脑用10%甲醛盐水固定,常规处理,用甲酚固紫(CFV)染色以显示尼氏物质,用胶质纤维酸性蛋白(GFAP)染色以显示星形胶质细胞的表达。CFV染色显示曲马多治疗组这些有病理改变的区域染色强度降低。GFAP显示大量反应性星形胶质细胞突起;星形胶质细胞突起重叠和交错;星形胶质细胞增殖;星形胶质细胞细胞体肥大和星形胶质细胞突起增厚。本研究结果揭示了重量、尼氏体和海马形成组织病理学的变化。关键词:海马形成、组织化学、组织病理学、免疫组织化学、神经变性引言阿片类药物滥用已成为一场全球健康危机,影响着世界各地不同背景和社区的个人。在阿片类药物中,曲马多已获得

飞行员和飞行员事故错误的航空医学原因
专家认为航空业的失误是导致事故和事件的主要因素。本文研究了导致尼日利亚飞行员和飞机工程师发生事件或事故的航空医学因素。本文利用了对随机抽样受访者进行问卷调查收集的数据。总共向飞行员和飞机工程师发放了 300 份问卷。使用因子分析和多元回归分析相结合的方式分析数据。因子旋转后提取的变量表明,一般健康状况(78.20%)是导致飞机工程师发生错误的最重要原因。对于飞行员而言,迷失方向(79.20%)被发现是导致错误的最关键的航空医学原因。多元回归分析结果显示,航空工程师的 R = 0.651,飞行员的 R = 0.607。这些发现表明,航空事故和由错误引起的事件可以追溯到这些航空医学因素。本文建议在航空专业人员的许可和重新认证指南中增加对航空医学条件的严格执行,以便将尼日利亚航空业中可追溯到错误的事故和事件减少到最低限度。

森林砍伐:原因以及欧盟如何应对它|新闻
气候变化既是森林砍伐和森林退化的原因,也是结果。它触发的极端事件,例如火灾,干旱和洪水,会影响森林。反过来,森林损失对气候有害,因为森林在提供清洁空气,调节水周期,捕获二氧化碳,防止生物多样性丧失和土壤侵蚀方面发挥了重要作用。欧盟在森林砍伐土地上生产的商品的消费

1 Eg5 抑制剂 SB743921 导致 p53 依赖性细胞...
。CC-BY-NC-ND 4.0 国际许可证永久有效。它以预印本形式提供(未经同行评审认证),作者/资助者已授予 bioRxiv 许可,可以在该版本中显示预印本。版权所有者于 2025 年 1 月 25 日发布了此版本。;https://doi.org/10.1101/2025.01.23.634373 doi:bioRxiv 预印本

AN1785,ESD 和 EOS 的原因、区别和预防
1.电源电压浪涌超出绝对最大电压范围。2.电路板上的开关电路可能会导致电路板内部出现高压尖峰,并传播到电路板上的其他设备。3.外部连接(例如外部电缆上的电容电荷、天线拾取的外部开关噪声和电感负载)可能会产生电压尖峰。4.由于接地不良导致接地平面上出现过大噪声。5.I/O 切换产生电压过冲或下冲。6.由于电气噪声环境中的屏蔽不良而产生 EMI(电磁干扰)。7.不正确的上电顺序可能会对设备施加非预期的电压水平或极性。8.ESD 事件会导致设备损坏或削弱设备,使其更容易受到未来 EOS 事件的影响。9.如果电流很高或持续时间很长,闩锁事件可能会导致 EOS 损坏。

原因,鉴定和治疗阿尔茨海默氏病
阿尔茨海默氏病是以遗传和环境因素为特征的最常见的神经退行性疾病之一。目前的工作基于与AD早期发作及其诊断意义相关的基因突变。本研究采用了定性研究方法,涉及对现有文献的分析,以确定PSEN1和PSEN2基因的遗传变异并比较当前的诊断选择。四个关键突变 - p。这些是Thr119ile,P.Gly209Ala,P.Gly417Ala和P.His169asn突变,与阿尔茨海默氏病早期发作有关。关于神经影像学和生物标志物的新研究在初始阶段提供了更好的诊断准确性。然后将这些遗传发现与其他环境和生活方式因素进行比较,以便显示两者之间的多方面关系与它们对疾病发展和进展的影响。对特定基因突变的识别可以提高AD病因的知识,并可以在实践中应用先进的诊断方法,这表明在AD治疗中进一步开发了个性化医学。
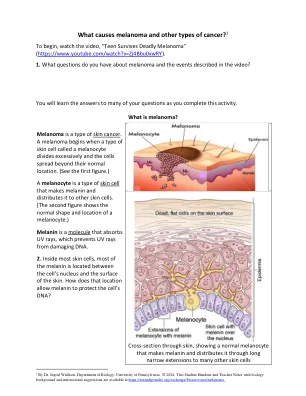
是什么引起黑色素瘤和其他类型的癌症?1
C.如果免疫细胞和手术没有消除所有黑色素瘤细胞,那么存活的黑色素瘤细胞可以在肺,肝或其他器官中建立转移性黑色素瘤。转移性黑色素瘤可以通过干扰其所在的器官的功能,并使用如此多的营养来杀死一个人,以至于人体其余部分的健康细胞无法获得足够的营养。4a。视频中的第一位青少年的第一位医生删除了她脸上的黑色素瘤。但是,他警告她,黑色素瘤可以在五年内杀死她。去除脸上的黑色素瘤后,黑色素瘤细胞在她体内可以在哪里?4B。黑色素瘤最终如何导致她的死亡?为了防止黑色素瘤可能杀死这名少年,她的第二位医生去除了淋巴结(如果它们含有黑色素瘤细胞)。他还开了药物,以帮助她的免疫细胞更有效地识别和破坏黑色素瘤细胞。

drsg 1 - 糖尿病的原因DRSG 2 - 类型1糖尿病
Areas of work: • Preventing harm in acute care • Machine learning to identify high-risk patients • Training schemes • Pregnancy and diabetes in inpatients • Covid-19's impact on inpatient diabetes care • Capacity building in inpatient diabetes research • Technology to support acute care • Glycaemic measurements and their use in acute care • PROMS

血液疾病:类型、原因、诊断和治疗
缺铁性贫血:最常见的贫血类型,由缺铁引起,而铁是产生血红蛋白所必需的。维生素缺乏性贫血:由缺乏维生素 B12 或叶酸引起,而这两种物质对于红细胞的产生至关重要。镰状细胞性贫血:一种遗传性疾病,红细胞呈新月形,会阻碍血液流动,导致疼痛和器官损伤。再生障碍性贫血:一种罕见疾病,骨髓无法产生足够的红细胞、白细胞和血小板。溶血性贫血:当红细胞被破坏的速度超过人体替换的速度时就会发生。贫血的症状包括疲劳、虚弱、皮肤苍白、呼吸急促和头晕。治疗取决于病因,可能包括补铁、注射维生素 B 12、输血或使用刺激红细胞产生的药物。

了解冠状动脉疾病:原因,症状...
冠状动脉疾病(CAD)是心脏病的最普遍形式之一,也是全球发病率和死亡率的主要原因。当冠状动脉向心肌提供血液时,由于脂肪沉积,胆固醇和其他物质的积聚而被狭窄或阻塞,统称为动脉粥样硬化。了解CAD对于预防,早期检测和有效的管理至关重要。CAD的开发是多因素的,具有各种危险因素。这些因素可以分类为可修改和不可修改的风险。低密度脂蛋白(LDL)胆固醇水平升高会导致动脉斑块积聚。高血压会损害动脉,使它们更容易受到动脉粥样硬化的影响。烟草使用是一个重要的危险因素,因为它会导致血管变窄并增加血压。胰岛素抵抗和高血糖水平会损害血管,从而促进CAD。过多的体重与较高的胆固醇水平,高血压和糖尿病有关,所有这些都导致了CAD。久坐的生活方式与肥胖症和心脏病的其他风险因素有关。饱和脂肪,反式脂肪,胆固醇和钠含量高的饮食可以促进CAD发育。CAD的风险随着年龄的增长而增加,特别是对于45岁以上的男性,超过55岁的女性通常在生活中的风险较早,尽管女性的风险增加了更年期。心脏病的遗传易感性可以提高个人的风险。[1,2]。