XiaoMi-AI文件搜索系统
World File Search Systemclones

高效 CRISPR/Cas9 介导全色盲患者来源的 iPSC 中纯合突变的校正
摘要:全色盲是一种常染色体隐性遗传病,患者视锥细胞会逐渐退化,导致色盲和视力下降,以及其他严重的眼部病变。它属于一类遗传性视网膜营养不良症,目前尚无治疗方法。尽管一些正在进行的基因治疗研究报告了功能改善,但仍应开展更多努力和研究以增强其临床应用。近年来,基因组编辑已成为个性化医疗最有前途的工具之一。在本研究中,我们旨在通过 CRISPR/Cas9 和 TALENs 技术纠正全色盲患者 hiPSC 中的纯合 PDE6C 致病变异。在这里,我们展示了 CRISPR/Cas9 的高基因编辑效率,但 TALENs 近似值不高。尽管少数经过编辑的克隆表现出杂合的靶向缺陷,但具有潜在恢复的野生型 PDE6C 蛋白的校正克隆的比例占所分析克隆总数的一半以上。此外,它们中没有一个出现脱靶畸变。这些结果对单核苷酸基因编辑的进展和未来治疗全色盲的策略的发展做出了重大贡献。

使用通用供体
图1。通过MESC中的刺激诱导的插入诱变。(a)击球策略的示意图。通过Cas9 RNP的hit-trap供体和基因组的同时裂解会导致靶向捕获。 整合后,基因陷阱盒会导致靶基因启动子的截短蛋白和GFP的表达。 选择盒子由组成型SV40启动子表达紫霉素的抗性基因。 ATS序列:GGTATGTCGGGAACCTCTCCAGG; SA,剪接受体; IRES,内部核糖体入口网站; PA,聚腺苷酸信号。 (b)在杀击球中选择呼吸霉素后MESC克隆的代表性微观图像。 红色箭头分别指示凋亡克隆(顶部),GFP-生存的克隆(中间)和GFP阳性幸存的克隆(底部)。 比例尺,50 µm。 (c)GFP阳性克隆的PCR基因分型证实了HPRT基因座的hit-trap供体的正确整合。 5 /3J,5' /3'交界处。 (d,e)针对HPRT基因座(TH1-1,TH2-4和TH3-5)的hit-trap克隆的Western印迹和QPCR分析,并用微管蛋白作为负载对照。 错误条显示了S.D. 来自三个技术重复。 使用学生的未配对t检验来计算显着性:** p <0.01。通过Cas9 RNP的hit-trap供体和基因组的同时裂解会导致靶向捕获。整合后,基因陷阱盒会导致靶基因启动子的截短蛋白和GFP的表达。选择盒子由组成型SV40启动子表达紫霉素的抗性基因。ATS序列:GGTATGTCGGGAACCTCTCCAGG; SA,剪接受体; IRES,内部核糖体入口网站; PA,聚腺苷酸信号。(b)在杀击球中选择呼吸霉素后MESC克隆的代表性微观图像。红色箭头分别指示凋亡克隆(顶部),GFP-生存的克隆(中间)和GFP阳性幸存的克隆(底部)。比例尺,50 µm。(c)GFP阳性克隆的PCR基因分型证实了HPRT基因座的hit-trap供体的正确整合。5 /3J,5' /3'交界处。(d,e)针对HPRT基因座(TH1-1,TH2-4和TH3-5)的hit-trap克隆的Western印迹和QPCR分析,并用微管蛋白作为负载对照。错误条显示了S.D.来自三个技术重复。使用学生的未配对t检验来计算显着性:** p <0.01。

干细胞研究
转基因株系采用第二代 CRISPRa 系统,该系统携带与异源三聚体 VPR 反式激活因子融合的核酸酶缺陷型 dCas9,该异源三聚体 VPR 反式激活因子由 VP64、p65 和 RTA 结构域组成。该系统可用于解释任何所需细胞类型的内源性调控机制。使用基于 CRISPR/Cas9 的基因组编辑方法,我们以 AAVS1 人类基因组位点为目标,分别引入先前描述的 dCas9VPR-tdTomato(Schoger 等人,2020 年)和嘌呤霉素盒,这些盒受 CAG 和 EF1a 启动子的控制(图 1 A)。采用优化的核转染方案转染 LhiPSC-GR1.1 细胞。转染后,选择具有 tdTomato 表达的细胞并通过 PCR 进行基因分型(图 1B,引物结合如图 1A 所示,黑色引物仅扩增野生型 (WT) 片段;绿色引物扩增插入的构建体)。随后,扩增、分析和冷冻保存两个阳性克隆(#2 和 #3)。DNA 测序数据证实了 AAVS1 基因座中的正确和纯合敲入转基因整合(图 1C,显示为克隆#2)。PCR 结果显示,在筛选的 15 个克隆中,11 个克隆含有纯合插入(命名为 CRISPRa 细胞),1 个克隆是杂合的,3 个克隆不含有插入而是含有 WT 完整基因座(用作对照细胞)(数据未显示)。通过分析 PCR 和测序预测的前五个脱靶位点进行脱靶分析;在这些位点中均未发现任何编辑事件。对照电穿孔和非电穿孔 (参考) 系用于比较 (补充图 1A)。所有系的支原体检测均为阴性。通过基于 SNP 的核型分析和标准 G 带证明了 CRISPRa 克隆 #2 和 #3 以及对照细胞的基因组完整性。未检测到数值或结构异常的证据 (图 1D)。与核转染 (图 1Ei) 和非核转染对照相比,细胞生长和形态正常。与对照 hiPSC 相比,CRISPRa 中的 dCas9 和 tdTomato 表达证实了转基因表达,如 Western blot (补充图 1B,显示克隆 #2 和 #3) 和共聚焦显微镜 (图 1Eii,显示克隆 #2,n = 3 个不同传代) 所示。通过免疫荧光分析干性标记 OCT4 的表达(图 1 Eiii)和流式细胞术分析(显示 94.2% OCT4 和 99.9% TRA1-60 阳性细胞(图 1 Eiv)(显示克隆 #2))来评估多能性。通过在 CRISPRa 和对照系中形成胚状体 (EB) 和定向分化来测试向所有三个胚层的自发分化能力。免疫荧光分析证实了 AFP、β-III-Tu bulin 和 α-平滑肌肌动蛋白 (ACTA2) 的表达,进一步支持内胚层、外胚层和中胚层的命运(图 1 F,显示克隆 #2 和 #3)。转录水平分析表明配对盒 3 ( PAX3 ) 和微管相关蛋白 2 ( MAP2 ) 的表达表明外胚层分化;T-box 转录因子 T ( TBXT ) 表明中胚层命运,而 α-Feto-Protein ( AFP ) 表明内胚层分化(补充图 1 C,显示克隆 #2 和 #3)。我们研究了 CRISPRa 系用于研究通过定向 2D 分化产生的心肌细胞的适用性,这种分化产生了自发跳动的细胞(视频作为补充材料提供),具有强大的 α-辅肌动蛋白 2 (ACTN2) 和心脏肌钙蛋白 T (TNNT2) 心脏标志物表达((补充图 1D,显示为克隆#2)。最后,我们通过确定与心脏肥大和代谢稳态有关的 KLF15 表达的诱导来测试 CRISPRa 系的功能。我们发现,与转染了非靶向 gRNA 的各自亲本系相比,设计用于结合 KLF15 转录起始位点 (TSS) 的 44 bp 5'-上游序列的单个指导 RNA 能够显着增强 CRISPRa 系(克隆#2 和#3)中 KLF15 的转录。对照细胞没有显示独立于转染的 gRNA 的活化(图 1G)。总之,使用完全表征的 hiPSC 系,我们生成了具有纯合靶向插入、正常核型和多能性的人类 CRISPRa 系,并显示出其激活
IPSC衍生的视网膜色素上皮... 亚硝胺效力预测 使用生成对抗网络在药物发现中提高毒理学 - 第二部分 2024-学术学院的企业和工业 - ... 整合机器学习和定量
25个健康克隆28患病(> 30 AMD,4白白,1 Joubert,2 STAT3,2 Stargardt,2 L-ord)由> 2个实验室和½打公司复制

估计葡萄种植生物多样性:客观定量的新指数
培养的葡萄藤品种数量减少以及托儿所可用的植物材料和克隆的多样性以及葡萄酒生产商使用的后果仍然是许多争论的主题。以更好地理解和更准确地定义不同情况下不同情况下的缺点或优势,我们试图开发适合葡萄藤的不同索引,以比较中性和客观的方式。这些指标可能会考虑不同的空间水平(世界,国家,地区,庄园和地块),并可能考虑到不同类别的植物材料,例如品种,克隆或根骨。也可以应用它们来量化某些标签或认证计划的生物多样性水平,以保证消费者。
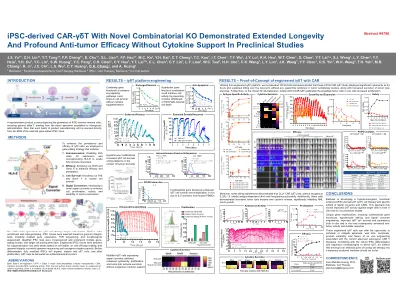
具有新型组合 KO 的 iPSC 衍生 CAR-γδT 在临床前试验中无需细胞因子支持即可表现出更长的寿命和显著的抗肿瘤功效
用于 iγδT 细胞疗法的 GMP 克隆生成始于人类 PBMC。在富集和重编程后,根据基因组完整性测试(包括残留基因表达、TCR 测序和形态学评估)选择 iPSC 克隆。合格的 iPSC 系被冷冻保存并经过多轮基因编辑,每轮之后进行单细胞分选。根据细胞健康、靶向和脱靶编辑以及基因组完整性(通过全基因组测序和致癌基因突变面板)选择工程 iPSC 克隆进行冷冻保存到种子库中。在分化之前,完全改造的 iPSC 将扩增、成熟为 γδT 细胞,增殖后,iγδT 细胞被收获为药品。

基于质粒的CRISPR敲入秀丽隐杆线虫
图1。基于质粒的CRISPR敲入的高度提高的克隆效率:(a)泳道1:NEB®DNA梯子标准(N3200S);泳道2:标准NEB®Q5PCR方案30个周期的Q5 PCR方案基于光涂抹和〜300bp的额外不需要的PCR产物导致过多的DNA,可重复出现30个周期。车道3-8:优化PDD162扩增的PCR循环编号。基于此数据,我们选择了15个周期作为PDD162所有后续扩增的最佳数字。(b)过多的PCR产物和DPNI消化不足会导致约35%的KLD连接克隆是错误的CAS9/SGRNA质粒。相反,优化PCR和KLD连接反应会导致90%的克隆具有正确的GRNA插入。(c)载体主链和用于

通过 CRISPR/Cas9 介导的同源重组将人类 gdnf 敲入牛 β-酪蛋白基因座*
帕金森病 (PD) 是一种常见且使人衰弱的神经退行性疾病,其源于多巴胺能神经元的损失,并伴有进行性运动功能障碍。神经胶质细胞衍生的神经营养因子 (GDNF) 在治疗 PD 和其他神经病方面非常有前景。在本研究中,我们应用 CRISPR/Cas9 技术开发了一种基因靶向敲入系统,用于在牛 β-酪蛋白基因位点表达人类 gdnf 基因。构建了 CRISPR/Cas9 表达质粒和 pP40-GN 载体。使用组织外植体法培养和收集牛胎儿成纤维细胞。然后将 pP40-GN 和 CRISPR/Cas9 载体电转染到牛胎儿成纤维细胞中。使用 G418 筛选抗性克隆,同时通过 PCR 分析和 PCR 产物测序鉴定目标克隆。采用耳组织阻断法成功分离培养牛胎儿成纤维细胞,将pP40-GN靶载体和CRISPR/Cas9表达载体共转染牛胎儿成纤维细胞,经7天G418筛选,共获得12个健康、分离良好的细胞克隆,其中5个发生基因打靶事件。本研究为利用基因打靶牛乳腺生物反应器生产人GDNF蛋白奠定了基础,为PD的靶向治疗提供了新的策略。

在 ES 细胞中进行 CRISPR 增强靶向后,靶向率和串联风险增加
摘要:法国小鼠诊所 (Institut Clinique de la Souris; ICS) 已生产出 2000 多个用于 C57BL/6N 小鼠“点菜”诱变的靶向载体。尽管大多数载体已成功用于小鼠胚胎干细胞 (ESC) 中的同源重组,但少数载体在多次尝试后仍无法靶向特定位点。我们在此表明,将 CRISPR 质粒与与之前失败的质粒相同的靶向构建体进行共电穿孔可以系统地获得阳性克隆。然而,必须仔细验证这些克隆,因为大量克隆(但不是全部)显示靶向质粒在位点处发生串联。详细的南方印迹分析可以表征这些事件的性质,因为标准的长距离 5′ 和 3′ PCR 无法区分正确和错误的等位基因。我们表明,在 ESC 扩增之前进行简单且廉价的 PCR 可以检测和消除带有串联体的克隆。最后,尽管我们只测试了小鼠 ESC,但我们的结果强调了任何结合使用 CRISPR/Cas9 和环状双链供体的转基因细胞系(如已建立的细胞系、诱导性多能干细胞或用于体外基因治疗的细胞系)存在错误验证的风险。我们强烈建议 CRISPR 社区在使用 CRISPR 增强任何细胞类型(包括受精卵母细胞)中的同源重组时,使用内部探针进行南方印迹。

细胞治疗 000 (2020) 113
背景:近年来,许多临床试验已成功测试了使用 CD4 + CD25 高 FoxP3 + 调节性 T 细胞 (Treg) 的疗法。在自身免疫性疾病中使用这种疗法的重要问题仍然是对特定抗原的特异性,因为由于表位扩散,在这种情况下通常有多种致病自身抗原需要调节。方法:我们在这里展示了一种生成富含抗原反应性克隆的 Treg 的方法,这种方法可能覆盖大多数此类自身抗原。在我们的研究中,用抗 CD28 和抗 CD154 抗体和自体单核细胞扩增 Treg,并装载模型肽,例如整个胰岛素或胰岛素 b 链肽 9 23。然后将细胞分选为识别呈递抗原的细胞。通过功能测定验证了反应性,其中 Treg 抑制了自体效应 T 细胞(多克隆和抗原特异性)的增殖或干扰素γ的产生,这些效应 T 细胞用作用模型肽攻击的应答者。最后,我们分析了特定 Treg 中的克隆型分布和 TRAV 基因的使用情况。结果:总之,应用的技术产量很高,并使我们能够获得富含特定亚群的 Treg 产品,这在功能测试中得到了证实。该产品由许多克隆组成;尽管如此,这些克隆的内容与多克隆或非特异性 Treg 中发现的内容不同。结论:所提出的技术可用于生成富含对任何给定肽有反应性的细胞的 Treg 群,可用作抗原靶向疗法中的细胞治疗药物。© 2020 国际细胞和基因治疗学会。由 Elsevier Inc. 出版。保留所有权利。