XiaoMi-AI文件搜索系统
World File Search Systemdystrophy

2025年1月27日|罗氏(Roche)宣布了对Duchenne肌肉营养不良的Elevidys的新结果|新闻| Chugai Pharmaceutical Co。,Ltd。
Delandistrogene Moxeparvovec,一种正在开发的基因治疗产品(开发法:SRP-9001,

与年龄相关黄斑变性的鉴别诊断
对年龄相关的黄斑变性(AMD)的诊断可能会对患者的生活产生重大影响。因此,考虑差异诊断是很重要的,因为这些诊断在预后,遗传,监测和治疗方面可能与AMD有很大差异。与drusen,类似drusen的变化,单基因视网膜营养不良以及许多其他罕见的黄斑疾病的AMD诊断有关其他黄斑疾病的差异诊断。在这篇综述中,提出了临床示例,以说明对AMD的替代诊断,以及何时应考虑这些诊断。These include, amongst others, patients with autosomal dominant drusen, Sorsby fundus dystrophy, pachydrusen, late-onset Stargardt disease, extensive macular atrophy with pseudodrusen (EMAP), pseudoxanthoma elasticum (PXE), North Carolina macular dystrophy, mitochondrial retinopathy, benign yellow dot黄斑病,圆顶或山脊形的斑块或黄斑telangiectasia类型2。

2024年先天性肌肉营养不良
Respiratory function in SEPN1- related myopathy and LAMA2 -related CMD ..................................................................................................................................... 13 Bone fragility in SEPN1- related myopathy and LAMA2 -related CMD ....... 13 Natural history of Dutch patients ............................................................................. 13 Outcome SEPN1的措施 - 相关的肌病和喇嘛与与CMD相关的CMD ....................................................................................................................................................................................................................................................................................... dystrophy - extracellular matrix ................................................. 15

神经肌肉疾病的运动功能测量量表……
针对神经肌肉疾病开发了一种新的运动功能测量量表。验证研究包括 303 名患者,年龄为 6-62 岁。72 名患者患有杜氏肌营养不良症,32 名贝克尔肌营养不良症,30 名肢带型肌营养不良症,39 名面肩胛肱骨营养不良症,29 名强直性肌营养不良症,21 名先天性肌病,10 名先天性肌营养不良症,35 名脊髓性肌萎缩症和 35 名遗传性神经病变。该量表包含 32 个项目,分为三个维度:站立姿势和转移、轴向和近端运动功能、远端运动功能。评分者间信度一致性系数在 9 个项目中为优秀 (k Z 0.81–0.94),在 20 个项目中为良好 (k Z 0.61–0.80),在 3 个项目中为中等 (k Z 0.51–0.60)。总分与其他分数之间存在高度相关性:Vignos (r Z 0.91) 和 Brooke (r Z 0.85) 等级、功能独立性测量 (r Z 0.91)、医生 (r Z 0.88) 和物理治疗师 (r Z 0.91) 使用视觉模拟量表评估的残疾总体严重程度。该量表可靠,不需要任何特殊设备,并且深受患者欢迎。正在评估其对变化的敏感性,以允许其用于神经肌肉疾病的临床试验。q 2005 Elsevier B.V. 保留所有权利。
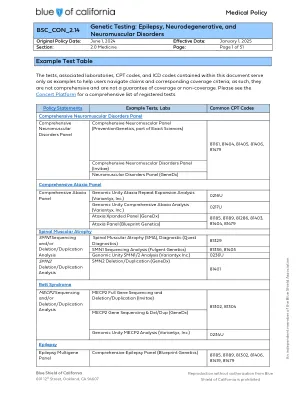
医疗政策BSC_CON_2.14基因测试:癫痫,...
1。具有一级或二级亲戚,对Duchenne或Becker肌肉营养不良的临床诊断。xxiii。dmd测序和/或缺失/重复分析(0218U,81161,81408)为所有其他适应症的研究都考虑了诊断Duchenne肌肉营养不良(DMD)或Becker肌肉营养不良(BMD)的诊断。facioscapulohumeral肌肉营养不良(FSHD)D4Z4单倍型分析和/或SMCHD1和DNMT3B测序和/或缺失/重复分析或Multigene Banel XXIV。D4Z4 haplotype analysis (81404), and/or SMCHD1 (81479) and DNMT3B (81479) sequencing and/or deletion/duplication analysis or multigene panel analysis (81404, 81479) to establish or confirm a diagnosis of facioscapulohumeral muscular dystrophy may be considered medically necessary when:

tilization M Edical P Olicy P Olicy:肌肉营养不良 - 基因疗法 - Elevidys利用管理管理医疗政策•Elevidys
•对于卧床患者,在DMD基因中具有确认的突变。•对于非注射性并在DMD基因中有确认突变的患者。基于骨骼肌中的levidys微肌营养蛋白的表达,非疗法患者的DMD指示得到了加速批准。在验证性试验中,持续批准了此指示可能取决于对临床益处的验证和描述。疾病概述DMD是由DMD基因突变引起的一种罕见的,进行性X连锁的疾病,也称为肌营养不良蛋白基因。2-4美国DMD的发病率约为5,000名活着的男性出生。 DMD基因是最大的已知人类基因,大小总计2.3兆瓦。 该基因编码功能性肌营养不良蛋白,该蛋白是跨膜蛋白复合物的一部分,跨膜蛋白复合物跨越了骨骼和心脏肌肉细胞的肌膜。 这种复合物将细胞骨架与细胞外基质联系起来,从而为肌膜提供结构完整性,并有助于传递和吸收与肌肉收缩相关的休克。 DMD基因中的突变可防止功能性肌营养不良蛋白或肌营养不良蛋白的产生。 没有肌营养不良蛋白,DMD患者的正常活性会对肌肉纤维细胞造成过度损害。 随着时间的流逝,肌肉细胞被脂肪和纤维化组织代替。 进行性肌肉无力是DMD的主要表现。 这会导致失去行动,相关运动延迟,呼吸障碍和心脏功能的逐步下降。2-4美国DMD的发病率约为5,000名活着的男性出生。DMD基因是最大的已知人类基因,大小总计2.3兆瓦。该基因编码功能性肌营养不良蛋白,该蛋白是跨膜蛋白复合物的一部分,跨膜蛋白复合物跨越了骨骼和心脏肌肉细胞的肌膜。这种复合物将细胞骨架与细胞外基质联系起来,从而为肌膜提供结构完整性,并有助于传递和吸收与肌肉收缩相关的休克。DMD基因中的突变可防止功能性肌营养不良蛋白或肌营养不良蛋白的产生。没有肌营养不良蛋白,DMD患者的正常活性会对肌肉纤维细胞造成过度损害。随着时间的流逝,肌肉细胞被脂肪和纤维化组织代替。进行性肌肉无力是DMD的主要表现。这会导致失去行动,相关运动延迟,呼吸障碍和心脏功能的逐步下降。DMD的第一个临床症状是运动发展里程碑的延迟,例如步行,这是2岁左右的观察到的。通常会延迟诊断直到3至5岁。年龄是DMD进展的重要预后因素。目前无法治愈DMD。治疗的目的是管理症状,缓慢的疾病进展并延迟残疾。患有DMD的男孩通常会失去12岁或13岁以上行走的能力。过去,死亡率是在青春期或二十年代初发生的,但是随着呼吸道和心脏管理的进步,有些患者居住到第四个十年。DMD患者最常见的死亡原因是呼吸衰竭,呼吸道感染,心肌病和心律不齐。皮质类固醇是DMD治疗的中流型;但是,其在DMD中的作用机理尚不清楚。皮质类固醇可以改善疾病的症状,并延迟流动和其他后遗症的时间。Four anti-sense oligonucleotide therapies (exon-skipping) have been approved by the FDA: Exondys 51 ® (eteplirsen intravenous infusion), Vyondys 53 ™ (golodirsen intravenous infusion), Viltepso ™ (viltolarsen intravenous infusion), and Amondys 45 ™ (casimersen intravenous输液)。由于没有完成任何确认性临床研究,因此这些外显子的疗法的临床益处仍然未知。临床疗效在三项研究中评估了leverdys的疗效:1-4,7-9 Engark III期随机,双盲,安慰剂对照,确认性试验; II期研究;和IB期研究。在Embark(n = 125)中,从基线到第52周的主要终点

我们在 FSHD 中处于什么位置以及我们是如何到达这里的?
FDA(2R01FD006071)、CDC(1U01DD001242)、强直性肌营养不良症基金会、肌肉萎缩症协会、钙蛋白酶 3 治疗联盟、Fulcrum、AMO Pharma、Sarepta、Dyne、Vertex、Edgewise、Novartis、ML Bio、辉瑞 • 顾问/顾问委员会:Sarepta、AskBio、Acceleron、
Roche神经病学更新虚拟IR事件2024年3月11日
1 bepranemab与UCB合作,目前由UCB进行研究; 2与Sarepta Therapeutics合作的Elevidys; NME =新分子实体; AI =其他指示; NMOSD =神经瘤性光谱谱系障碍; DMD = Duchenne肌肉营养不良; GMG =广泛的肌腱肌症; SMA =脊柱肌肉萎缩; fshd = facioscapulohumeral肌肉营养不良; mog-ad =髓磷脂少突胶质细胞糖蛋白抗体相关疾病; AIE =自身免疫性脑炎; MAGL =单酰基甘油脂肪酶


审议结果报告
1. 发现的来源或历史、在国外的使用及其他信息 杜氏肌营养不良症(DMD)是一种X连锁隐性遗传病。该病是由X染色体上的肌营养不良蛋白基因突变缺失或重复导致功能性肌营养不良蛋白缺陷引起的(Cell. 1987;51:919-28)。DMD是“肌营养不良症”的一种指定难治性疾病,是一种难治性进行性肌肉疾病,并发呼吸肌和心肌无力以及严重的运动功能障碍、吞咽困难、痰液滞留和胃肠道功能障碍。患有 DMD 的儿童在 10 岁左右失去行走能力,平均寿命约为 30 年(杜氏肌营养不良症 (DMD) 实用指南 2014。Nankodo Co., Ltd.;2014:2-5)。每 3500 名新生男婴中就有 1 名患有 DMD(Neuromuscul Disord. 1991;1:19-29),估计日本约有 5000 名患者受到影响(Experimental Medicine. 2016;34:3151-8)。鉴于大约 8% 的 DMD 患者具有可使用 viltolarsen 治疗的基因突变(Hum Mutat. 2009;30:293-9),预计日本约有 400 名患者可使用 viltolarsen。 2019 年 8 月 20 日,Viltolarsen 被指定为孤儿药(孤儿药指定编号 2019 年第 440 号 [ 31 yaku ]),预期适应症为“杜氏肌营养不良症,肌营养不良蛋白基因缺失,可通过外显子 53 跳跃疗法治疗”。Viltolarsen 是一种合成的吗啉寡核苷酸,由申请人和美国国家神经病学和精神病学中心开发。Viltolarsen 与肌营养不良蛋白信使核糖核酸 (mRNA) 前体的外显子 53 结合,从而跳过外显子 53,导致肌营养不良蛋白的表达,这种蛋白比正常蛋白链短,但具有功能性。在日本,2013年6月,由国立神经精神病学中心以厚生劳动科学研究基金资助的研究者发起试验的形式开始了临床研究。申请人提交了viltolarsen的上市申请,声称viltolarsen在DMD患者中已证实了其有效性和安全性。在美国,viltolarsen的申请于2019年12月提交,目前正在审查中。截至2019年12月,viltolarsen尚未在任何国家或地区获得批准。日本批准的肌营养不良症适应症药物有泼尼松龙(适应症为“杜氏肌营养不良症”)和三磷酸腺苷二钠水合物注射剂(适应症为“肌营养不良症及相关疾病”)。 Viltolarsen 于 2015 年 10 月 27 日被指定为 SAKIGAKE 指定系统的对象(2015 年 SAKIGAKE 药品指定第 2 号 [ 27 yaku ]),其预期适应症为“杜氏肌营养不良症”。“Viltolarsen 还受到药品有条件早期批准制度的约束(PSEHB/PED 通知编号 1029-3,2019 年 10 月 29 日)。