XiaoMi-AI文件搜索系统
World File Search SystemeQTL

利用已建立的基于生物信息学的方法,实现基因组变异驱动的结核病药物再利用
将结核病 (TB) 基因组变异转化为临床应用的一个主要挑战是整合与疾病相关的变异,并通过基因组驱动的药物再利用概念促进药物发现。在这里,我们利用两个已建立的基因组数据库,即全基因组关联研究 (GWAS) 和全表型关联研究 (PheWAS) 来识别与结核病相关的基因组变异,并进一步将其用于药物靶向基因。我们评估了 3.425 个与结核病相关的基因组变异,这些变异与 200 个结核病相关基因重叠。为了确定生物结核病风险基因的优先级,我们设计了一个计算机模拟流程,并利用基于六个功能注释(错义突变、顺式 -eQTL、生物过程、细胞成分、分子功能和 KEGG 分子通路分析)的成熟生物信息学方法。有趣的是,基于我们应用的六个功能注释,我们发现 14 个生物结核病风险基因与生物结核病风险基因的失调密切相关。因此,我们证明了 12 个药物靶基因与 40 种用于其他适应症的药物重叠,并进一步表明这些药物可能被重新用于治疗结核病。我们强调 CD44、CCR5、CXCR4 和 C3 是极有前途的拟议结核病靶点,因为它们与 SELP 和 HLA-B 相关,而 SELP 和 HLA-B 是功能注释上系统评分较高的生物结核病风险基因。总之,当前的研究揭示了与结核病发病机制有关的基因组变异作为生物结核病风险基因,并提供了结核病基因组学可能有助于药物发现的经验证据。

人类胰岛胰岛microRNA与大规模遗传分析有关的糖尿病和相关性状
遗传研究已经确定了与2型糖尿病风险相关的≥240个基因座(T2D),但这些基因座大多数位于非编码区域,掩盖了基本的分子机制。最近研究人类胰岛中mRNA表达的研究已经对正常胰岛功能和T2D病理生理学的分子驱动因素产生了重要的见解。但是,研究microRNA(miRNA)表达的类似研究仍然有限。在这里,我们提供了来自63个个体的数据,这是迄今为止人类胰岛中miRNA表达的最大测序分析。我们通过将高度可遗传的miRNA的表达分解为顺式和转移的遗传成分,并映射与miRNA表达相关的顺式基因座[miRNA表达定量性状特质基因座(EQTLS)]。我们发现I)84可遗传的miRNA,主要由反式遗传效应调节,ii)5 miRNA -EQTL。我们还使用了几种不同的策略来识别与T2D相关的miRNA。首先,我们将miRNA-EQTL与与T2D和多个血糖性状相关的遗传基因座共定位,鉴定了一种miRNA miRNA,miRNA,MiRNA,该miRNA共享血糖和糖化血糖的遗传信号(HBA1C)。接下来,我们将miRNA种子区域和与T2D和血糖性状相关的可靠的SNP相交,并发现了32个miRNA,这些miRNA可能因种子区域而破坏的结合和功能可能改变。最后,我们进行了差异表达分析,并确定了与T2D状态相关的14个miRNA,包括miR-187-3p,miR-21-5p,miR-668和miR-199b-5p,以及与HbA1C水平的多基因分数相关的4个miRNA,MIR-216A,mir-25,mir-25,mir-25,mir-30a-30a-3p和mir-3p和mir-3p和mir-3p。

ASEP:通过 RNA 测序对群体中个体间等位基因特异性表达进行基因检测
等位基因特异性表达 (ASE) 分析可量化二倍体个体中两个等位基因的相对表达,是识别顺式调控基因表达变异的有力工具,而顺式调控基因表达变异是个体间表型差异的基础。现有的基因水平 ASE 检测方法每次仅分析一个个体,因此无法解释个体间共享的信息。无法容纳这种共享信息不仅会降低检验能力,而且难以解释个体间的结果。然而,当只有 RNA 测序 (RNA-seq) 数据可用时,跨个体的 ASE 检测具有挑战性,因为数据通常包括未观察到的顺式调控 SNP 杂合或纯合的个体,从而导致样本异质性,因为只有杂合个体才对 ASE 具有信息性,而纯合个体的表达则均衡。为了同时对多个个体的信息进行建模并解释这种异质性,我们开发了 ASEP,这是一种具有受试者特定随机效应的混合模型,用于解释同一基因内的多 SNP 相关性。ASEP 只需要 RNA 测序数据,并且能够检测一种条件下的基因水平 ASE 和两种条件(例如,治疗前和治疗后)之间的差异 ASE。广泛的模拟证明了 ASEP 在各种情况下的令人信服的性能。我们将 ASEP 应用于人类肾脏 RNA 测序数据集,识别出 ASE 基因,并通过两项已发表的 eQTL 研究验证了我们的结果。我们进一步将 ASEP 应用于人类巨噬细胞 RNA 测序数据集,识别出显示 M0 和 M1 巨噬细胞之间存在差异 ASE 证据的基因,并通过心脏代谢特征相关的全基因组关联研究的结果证实了我们的发现。据我们所知,ASEP 是第一种仅需使用 RNA 测序数据即可在人群水平上进行基因水平 ASE 检测的方法。随着 RNA-seq 的日益普及,我们相信 ASEP 将非常适合针对人类疾病的各种 ASE 研究。

系统性可用药物的全基因组孟德尔随机化确定了慢性牙周炎的治疗靶基因
4 5 1 遵义医科大学附属口腔医院口腔颌面外科,贵州省遵义市。7 2 遵义医科大学口腔医学院、口腔医院,贵州省遵义市。8 3 遵义医药高等专科学校信息技术网络管理中心,贵州省遵义市。10 11 # 这些作者对本文贡献相同。12 ‡ 这些作者对本文贡献相同。13 * 通讯作者:宋庆高 (814641639@qq.com)14 15 摘要16 目前,慢性牙周炎 (CP) 的治疗仍面临挑战。本研究旨在利用孟德尔随机化(MR)方法在可用药物基因组中鉴定出治疗慢性牙周炎的新药物靶点。在本研究中,基于 4479 个药物基因靶点列表,在血液表达数量性状位点(eQTL)中选择了重叠基因,然后对其进行双样本 MR(TSMR),并使用来自 CP 的全基因组关联研究(GWAS)数据验证了融合转录组关联研究(TWAS),以确认与 CP 遗传相关的药物基因,并使用基于汇总数据的孟德尔随机化(SMR)分析和共定位测试多重效应。最后,对确定的药物靶点进行表型组关联研究(PheWAS)。我们应用SMR、TSMR、Fusion TWAS和一系列共定位方式来评估可用药靶点与CP之间的遗传关联。综上所述,金属蛋白酶25(MMP25)被认为是最有前途的药物靶点,此外,我们对TNFRSF18、CDC25B、STK10和ACVR2B也抱有一定信心。最后,PheWAS-MR结果显示,应用MMP25抑制剂的可能副作用包括外耳炎以及一些代谢紊乱等。综上所述,我们利用TSMR、Fusion TWAS、SMR和共定位等一系列方法确定了五个潜在的CP药物靶点,其中MMP25通过了所有测试。这一发现为未来CP药物开发提供了学术基础,并在一定程度上缩短了药物开发时间和经济成本。关键词:慢性牙周炎,全基因组孟德尔随机化,靶基因。
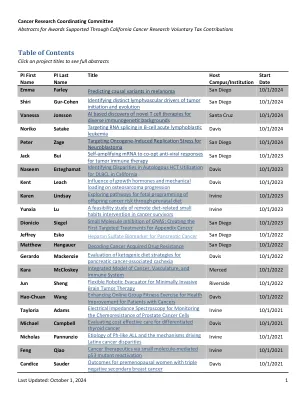
目录
预测黑色素瘤的致病变异 校园:UCSD 首席研究员:Emma Farley 开始日期:2024 年 10 月 1 日 结束日期:2025 年 9 月 30 日 金额:95,000 美元 摘要:癌症的各个方面都由基因表达的变化驱动。增强子是控制特定基因或基因表达的时间、位置和水平的基因组元素,因此,增强子提供基因表达的指令。虽然人们广泛关注蛋白质编码变异和改变蛋白质编码区域的基因组变化,但增强子变异如何促进癌症的发生、进展、转移和对治疗的反应却研究甚少。增强子包含与包括癌症在内的疾病相关的大多数变异,但精确定位致病变异是一项重大挑战,因为它们通常嵌入在大量惰性变异中。我们知识上的这一空白阻碍了充分利用基因组数据的潜力来了解和治疗癌症的努力。清楚了解哪些增强子变异对癌症的各个方面有影响对于了解癌症发生和发展的遗传基础、开发新型疗法、改善诊断和对患者进行分层以进行更有针对性的治疗至关重要。在这项研究中,我们将试行一种新方法来识别导致黑色素瘤基因表达变化的增强子变异。我们是识别增强子中因果变异的专家,这些变异会在发育中的胚胎中改变基因表达和细胞身份。我们发现,低亲和力结合位点对于精确控制基因表达至关重要。增强子中低亲和力位点的普遍使用造成了基因组中的脆弱性,SNV 可以增加增强子中结合位点的亲和力,从而导致功能基因表达获得,从而改变细胞身份。我们已经在心脏和肢体发育的背景下证明了这一点。我们现在希望应用这些知识来深入了解导致癌症各个方面的因果变异。我们计划最初重点关注黑色素瘤的转移、免疫疗法反应和耐药性。在我们的初步研究中,我们已表明,在体细胞黑色素瘤 eQTL 中发现的亲和力优化 SNV 会增加 DAAM1 的表达并增加细胞迁移。成功完成该项目将揭示增强子变体对癌症进展和治疗的贡献。

揭开ADHD:基因,同时发生的性状和发展动力学
注意力缺陷/多动症(ADHD)是一种异质性神经发育状况,同事疾病的患病率很高,导致长期管理的难度增加。全基因组关联研究已经确定了多动症与共同出现的精神疾病之间共享的变异。但是,遗传机制尚未完全理解。这项研究将基因表达和空间组织数据纳入了胎儿和成人皮质组织中预测的因果ADHD基因的两样本的孟德尔随机化研究。他们鉴定了四个基因在皮质组织中的ADHD因果关系(胎儿:ST3GAL3,PTPRF,PIDD1;成人:ST3GAL3,TIE1)。蛋白质 - 蛋白质相互作用数据库与因果ADHD基因播种的生物学途径,将这些基因与条件联系起来(例如类风湿关节炎)和生物标志物(例如淋巴细胞计数)已知与ADHD相关,但没有先前显示的遗传关系。在成年肝组织上重复进行分析,在成年肝组织中,预定的因果ADHD基因ST3GAL3与胆固醇性状相关。研究得出的结论是,该分析提供了对ADHD,同时存在性状和生物标志物之间组织依赖性的时间关系的见解。重要的是,它提供了先前研究和未研究的同时存在条件之间的遗传相互作用的证据。该研究对该领域的关键贡献是,鉴定ADHD的促成因果关系的基因,并以组织和时间依赖性的方式显示其生物学途径如何与共同存在的性状和生物标志物联系起来。评论,这是一项真正令人印象深刻的综合研究,使用了高级方法,及时且描述了,具有多种优势,作者也详细介绍了一些局限性。值得突出显示并在下面进行详细介绍。作者在研究ADHD的基本遗传学方面的重要工作受到赞扬,该遗传学的遗传力为74%,大约三分之一的这种遗传力来自普通变体。是针对其他复杂多基因性状的观察到的,许多已鉴定的SNP都在基因组的非编码区域及其对ADHD发展和与共同发生性状的相互作用的影响(S)尚不清楚。因此,在这项研究中,作者询问了一个问题,即他们是否可以通过将它们整合到胎儿和成人脑特异性染色质染色质相互作用和EQTL数据的分析中来识别因果ADHD基因,以便理解为发展ADHD及其共同性性状的个人风险。

GATM基因中富含血统的变体与血清肌酐水平升高有关
抽象的背景血清杂星(SCR)水平用于使肾脏肾和健康。与其他祖先背景的人相比,非洲血统的个体具有更高的SCR水平 - 控制年龄,性别,大小,肾脏Funceon和疾病状况的差异。这种差异的原因是未知的。我们假设在非洲血统群体(非洲血统富含的变体)中可能存在相对高频的基因变种,与SCR级别升级的非洲血统个体有关。方法我们的研究样本由我们所有人的研究计划中的一个入门人组成。我们使用整个基因组序列数据为我们所有人的所有对象都进行了基因组序列数据,并选择了18,979个对非洲欧洲混合物,可用的SCR水平量度和人口统计协变量的18,979个对象。我们对此类群体进行了一系列对SCR水平的祖先信息的协会研究,以检验我们对与SCR相关的非洲祖先富含变体的假设。结果研究的入围室平均为80.8%的非洲和17.5%的欧洲血统。对骨scr的水平与女性的非洲血统(𝜌 = 0.79)和雄性(𝜌 = 0.84)有可能相关。使用标准GWA,基于单倍型的混合映射和祖先特定的GWAS,在15染色体(15Q223:45.3MB-45.5MB)上鉴定了同样的全基因组显着性相同的峰值。精细的映射IDENEFINFINFINFIEN,与GATM基因共同划分的14个变体的可信度集,该变体编码了一种生物合成酶Creaene,Creaene是Creaenine的代谢前体。铅GWAS变体的替代等位基因(RS2467850,CHR15:45379909:C:T)与SCR水平(β= 0.07,p = 2.28×10 -17)相关,与欧洲(0.413)相比,在较高的频率(0.413)中发现了(0.413)组(0.413)。这些变体中的13种与GATM表达相关,这是基于非洲裔美国人全血EQTL的先前研究,并且它们都显示出非洲血统增强的类似pamerns。基于10种非洲血统的变体的SCR多基因评分完全使观察到的非洲血统的scr级别与SCR水平相提并论。结论我们的发现表明,非洲血统的变体上调了GATM,从而解释了在非洲血统个体中观察到的较高水平的SCR,并强调了使用Geneec数据来衡量基因的poteneal caribibrate caribibrate caribibrate caribibrate newney funceon equaeone equaeon。关键词祖先,混合物,克雷氨酸,肾脏,全基因组协会研究(GWAS)