XiaoMi-AI文件搜索系统
World File Search Systemsyngeneic

CRISPR 筛选是否能提供下一代治疗靶点? Francisca Vazquez 1 和 William R. Sellers 1,2,3
精准癌症医学的成功取决于我们能否发现并针对患者肿瘤中的特定弱点进行治疗。理想情况下,我们将确定人类癌症中的所有靶点弱点,即维持癌症活力的靶点组合,并开发药物来抑制每个靶点。最后,我们需要非交叉耐药性治疗组合来克服人类肿瘤中发现的潜在亚克隆异质性。我们距离这一目标还很远,因为我们不完全了解癌症的弱点,而且我们缺乏针对大多数已识别弱点的药物。现在有两项进展使得在识别所有癌症弱点方面取得更大进展成为可能。首先,大量癌细胞模型的可用性和特性,虽然这些模型仍不完整,但可以开始模拟人类癌症的多样性。在这里,癌细胞系百科全书显著改变了在大量高度表征的细胞系 (1-3) 中分析治疗活性的能力。该集合现在已注释了包含 1,700 多个细胞系的数据集(可在 depmap.org 上获取)。模型生成方面的最新进展应该可以大大扩展这种多样性。第二次革命是在没有小分子抑制剂的情况下诱导基因特异性功能丧失的能力。此外,随着基因组规模 CRISPR 筛选的出现,现在可以以汇集的形式有效地改变每个基因的功能。CRISPR 筛选现在在大量细胞系或同源细胞系对中体外常规进行,在同基因小鼠模型中体内常规进行(4-6)。这些方法已经确定了正在药物发现计划中或新抑制剂处于临床开发中的治疗靶点(图 1)。也许同样重要的是现在能够确定特定疗法的更广泛有效性或无效性

对肿瘤淋巴瘤激酶(ALK)的药理抑制剂(ALK)通过靶向效应诱导免疫原性细胞死亡 基于注意力预测分子特性的定向消息传递 alpha活性神经调节由基于单个α的神经反馈学习在生态环境中诱导的:一项双盲随机研究 抗激素药物发现和发育:以所有可能的方式针对病毒及其宿主 基于大脑连接性的预测组合遥控器... 表皮生长因子受体的患者的存活... 大脑按时:发育与神经变性之间的联系 阿尔茨海默氏病中的β-分泌酶Bace1 人的大脑通过躯体镶嵌的镜头 认知时间的大脑信号箭头 人类神经干细胞的自我更新和分化之间的时间平衡需要淀粉样蛋白前体蛋白 全基因组对大肠杆菌中替代性肽聚糖交联所需的基因的鉴定揭示了-lactams的意外影响 多层网络中的遗传合作性暗示了亨廷顿氏病小鼠与症状同步的纹状体中的细胞存活和衰老
免疫原性细胞死亡(ICD)在临床上具有相关性,因为通过ICD杀死恶性细胞的细胞毒素会引起抗癌免疫反应,从而延长了化学疗法的影响,而不是治疗中断。ICD的特征是一系列刻板的变化,增加了垂死细胞的免疫原性:钙网蛋白在细胞表面的暴露,ATP的释放和高迁移率组Box 1蛋白以及I型Interferon反应。在这里,我们研究了抑制肿瘤激酶,间变性淋巴瘤激酶(ALK)的抑制可能性,可能会触发ICD在染色体易位因染色体易位而激活ALK的变性大细胞淋巴瘤(ALCL)中。多种证据辩称,有利于克唑替尼和塞替尼在ALK依赖性ALCL中的特异性ICD诱导作用:(i)它们在药理学相关的低浓度上诱导ICD Stigmata; (ii)可以通过ALK敲低模仿其ICD诱导效应; (iii)在支配碱性突变体的背景下失去了效果; (iv)通过抑制ALK下游运行的信号转导途径来模仿ICD诱导效应。当将经CERITIN的鼠类碱性ALCL细胞接种到免疫能力合成小鼠的左侧时,它们诱导了一种免疫反应,从而减慢了植入在右孔中的活Alcl细胞的生长。尽管Ceritinib诱导淋巴瘤小鼠的肿瘤的短暂收缩,无论其免疫能力如何,在免疫降低效率的背景下,复发频率更高,从而降低了Ceritinib对生存率的影响大约50%。完全治愈仅发生在免疫能力的小鼠中,并赋予了与表达同一碱性淋巴瘤的保护,但不与另一种无关的淋巴瘤进行保护。此外,PD-1阻滞的免疫疗法往往会提高治愈率。总的来说,这些结果支持了以下论点,即特异性ALK抑制作用通过诱导ICD诱导ALK-阳性ALCL刺激免疫系统。

Tigit-CD226-PVR轴:癌症免疫疗法的免疫检查点阻滞靶向抗体药物偶联物的抗中皮素会诱导癌症的抗肿瘤,并在癌症的小鼠模型中点燃抗肿瘤免疫力
抽象的背景新兴证据表明,化学疗法诱导的细胞死亡的机制可能会影响癌症患者的抗肿瘤免疫反应。与免疫学上无声的凋亡不同,凋亡是一种裂解和炎症形式的程序性细胞死亡,其特征是细胞膜中的孔形成和促炎性因子的释放。Gasdermin E(GSDME)最近通过某些化学治疗剂裂解GSDME后引起了人们的关注。这项研究研究了乳腺癌和结肠癌小鼠模型中,间皮素靶向抗体共轭物(ADC)的免疫调节作用。方法在EMT6乳腺癌和CT26结肠癌合成小鼠模型中研究了ADC的抗肿瘤作用。使用流式细胞仪分析ADC的免疫调节作用通过分析肿瘤浸润的免疫细胞。ADC作用机理。最后,在表达GSDME的肿瘤以及GSDME溶解的肿瘤中评估了ADC和FMS样酪氨酸激酶3配体(FLT3L)联合疗法的抗肿瘤作用。结果数据表明,ADC控制肿瘤的生长和刺激抗癌免疫反应。对作用机理的研究表明,微管,ADC的细胞毒性有效载荷,诱导GSDME的裂解以及诱发GSDME表达细胞中的凋亡细胞死亡。使用GSDME KO,我们表明GSDME表达对于ADC作为单一疗法的有效性至关重要。将ADC与FLT3L(一种细胞因子)结合在一起,该细胞因子在淋巴样和非淋巴组织中都扩展了树突状细胞,恢复了对GSDME KO肿瘤的控制。结论在一起,这些结果首次表明微管蛋白和含有ADC的微管蛋白会引起凋亡,并且这种烈性细胞死亡对于抗肿瘤的免疫和治疗反应至关重要。

RAGE 轴是右侧结肠免疫逃避的基础......
摘要 背景 右侧和左侧结肠的肿瘤发生具有不同的特征。目的 我们旨在描述代表结肠肿瘤发生早期的左侧和右侧腺瘤 (AD) 之间的差异。设计 分析单细胞和空间转录组数据集以揭示右侧和左侧结肠 AD 之间的改变。使用细胞、动物实验和临床标本来验证结果。结果 单细胞分析显示,在右侧 AD 中,杯状细胞显著减少,并且这些杯状细胞功能失调,粘蛋白生物合成减弱,抗原呈递缺陷。粘液屏障受损导致隐窝中形成生物膜,随后细菌侵入右侧 AD。空间转录组学显示,在隐窝周围有生物膜占据的区域经历了脂多糖 (LPS) 的炎症反应和细胞凋亡过程。在右侧 AD 中发现了独特的 S100A11 + 上皮细胞群,其表达水平由细菌 LPS 和肽聚糖诱导。S100A11 表达促进了同基因免疫功能正常小鼠的肿瘤生长,髓系抑制细胞 (MDSC) 增加,但细胞毒性 CD8+ T 细胞减少。用耐受性良好的晚期糖基化终产物 (RAGE) 受体拮抗剂 (Azeliragon) 靶向 S100A11 可显著抑制肿瘤生长和 MDSC 浸润,从而提高抗程序性细胞死亡蛋白 1 治疗结肠癌的疗效。结论我们的研究结果表明,功能失调的杯状细胞和随之而来的细菌易位激活了右侧结肠 AD 中的 S100A11-RAGE 轴,从而募集 MDSC 来促进免疫逃避。Azeliragon 靶向该轴可提高结肠癌免疫治疗的疗效。

海报#1
响应多种细胞信号,丝裂原活化蛋白激酶 MAP3K1 参与各种癌症信号网络,包括 NF κ B、JNK、ERK 和 p38 通路。MAP3K1 作为这些致癌通路中的信号激酶,促进肿瘤生长和转移。此外,胰腺癌患者中较高的 MAP3K1 转录水平与较差的 5 年生存率(50% vs. 15%)相关,这表明 MAP3K1 是癌症的一个有吸引力的治疗靶点。我们最近报道了一种喹喔啉类似物作为选择性 MAP3K1 抑制剂的发现(2022,PNAS)。使用 MAP3K1 AlphaFold 和 Schrödinger GLIDE 进行结构引导设计,得到 51-106,预计通过形成正交多极相互作用,它对 MAP3K1 的亲和力会提高。使用 KiNativ TM 平台在细胞基质中分析 51-106 表明 51-106 确实是一种具有改进效力的选择性 ATP 竞争性 MAP3K1 抑制剂。后续研究表明 51-106 阻断了 TNF α 诱导的 MAP3K1-IKK β 介导的 NF κ B 活性。51-106 抑制 MAP3K1 后进行的磷酸化蛋白质组学分析显示 NPM1 T199 磷酸化呈剂量依赖性下降,表明 NPM1 是 MAP3K1 的新底物。NPM1 在 DNA 损伤修复中起着关键作用;我们持续观察到 51-106 抑制 MAP3K1 后剂量依赖性的 S 期停滞,表明 DNA 损伤反应功能失调。用 MAP3K1 抑制剂 51-106 治疗胰腺癌细胞系可抑制细胞生长和迁移。在联合研究中,51-106 与吉西他滨在体外 LSL- KrasG12D/+、LSL-Trp53R172H/+、Pdx1-Cre (KPC) 细胞系和体内 KPC 同源原位移植小鼠胰腺癌模型中协同抑制生长。总之,我们使用结构引导设计开发了改进的 MAP3K1 抑制剂。我们的研究首次将 NPM1 确定为 MAP3K1 信号传导的成员,这些结果值得研究 MAP3K1 抑制作为癌症治疗选择。

靶向PPAR -GAMMA抵消适应...
抽象客观治疗诱导的肿瘤微环境(TME)重塑为癌症治疗带来了一个主要障碍。作为大多数肝细胞癌(HCC)患者表现出对反编程细胞死亡(配体)-1(抗PD- [L] 1)疗法的原发性或获得性的抗性,我们旨在研究对免疫接收靶标进行肿瘤适应的基础机制。设计通过抗PD-L1治疗的合成元素,免疫能力小鼠对HCC细胞的串行原位植入产生了两种抗免疫疗法的HCC模型,并通过单细胞RNA测序(SCRNA-SEQ),基因组和免疫分析对单细胞RNA测序(SCRNA-SEQ)进行询问。通过慢病毒介导的敲低和药理学抑制研究了关键信号通路,并通过对Pembrolizumab(NCT03419481)的II期试验进行了对HCC肿瘤活检的SCRNA-SEQ分析进一步验证。在没有明显的遗传变化的情况下,抗PD-L1耐药性肿瘤在免疫能力但不受免疫功能障碍的小鼠中比父母肿瘤大10倍,而这些小鼠的肿瘤变化伴随着髓样衍生的抑制细胞(MDSC)的肿瘤内积累(MDSC),cytotoxic cd8 + T细胞的细胞毒素和DESBORISECONS。从机械上讲,过氧化物酶体增殖物激活的受体伽马(PPARγ)转录活化活化的血管内皮生长因子-A(VEGF-A)产生以驱动MDSC扩张和CD8 + T细胞功能障碍的转录激活的血管内皮生长因子-A(VEGF-A)的产生。选择性的PPARγ拮抗剂触发了原位和自发性HCC模型中的免疫抑制至刺激性TME转化率,并将肿瘤变成抗PD-L1治疗。重要的是,对pembrolizumab抗性的HCC患者有40%(6/15)表现出肿瘤的PPARγ诱导。此外,较高的基线PPARγ表达与多种癌症类型的1例治疗患者的抗PD-(L)生存率较差有关。结论我们发现了一个适应性转录程序,肿瘤细胞通过PPARγ /VEGF-A介导的靶向免疫检查点靶向< /div < /div < /div

伽玛抵消适应免疫的肿瘤
抽象客观治疗诱导的肿瘤微环境(TME)重塑为癌症治疗带来了一个主要障碍。作为大多数肝细胞癌(HCC)患者表现出对反编程细胞死亡(配体)-1(抗PD- [L] 1)疗法的原发性或获得性的抗性,我们旨在研究对免疫接收靶标进行肿瘤适应的基础机制。设计通过抗PD-L1治疗的合成元素,免疫能力小鼠对HCC细胞的串行原位植入产生了两种抗免疫疗法的HCC模型,并通过单细胞RNA测序(SCRNA-SEQ),基因组和免疫分析对单细胞RNA测序(SCRNA-SEQ)进行询问。通过慢病毒介导的敲低和药理学抑制研究了关键信号通路,并通过对Pembrolizumab(NCT03419481)的II期试验进行了对HCC肿瘤活检的SCRNA-SEQ分析进一步验证。在没有明显的遗传变化的情况下,抗PD-L1耐药性肿瘤在免疫能力但不受免疫功能障碍的小鼠中比父母肿瘤大10倍,而这些小鼠的肿瘤变化伴随着髓样衍生的抑制细胞(MDSC)的肿瘤内积累(MDSC),cytotoxic cd8 + T细胞的细胞毒素和DESBORISECONS。从机械上讲,过氧化物酶体增殖物激活的受体伽马(PPARγ)转录活化活化的血管内皮生长因子-A(VEGF-A)产生以驱动MDSC扩张和CD8 + T细胞功能障碍的转录激活的血管内皮生长因子-A(VEGF-A)的产生。选择性的PPARγ拮抗剂触发了原位和自发性HCC模型中的免疫抑制至刺激性TME转化率,并将肿瘤变成抗PD-L1治疗。重要的是,对pembrolizumab抗性的HCC患者有40%(6/15)表现出肿瘤的PPARγ诱导。此外,较高的基线PPARγ表达与多种癌症类型的1例治疗患者的抗PD-(L)生存率较差有关。结论我们发现了一个适应性转录程序,肿瘤细胞通过PPARγ /VEGF-A介导的靶向免疫检查点靶向< /div < /div < /div
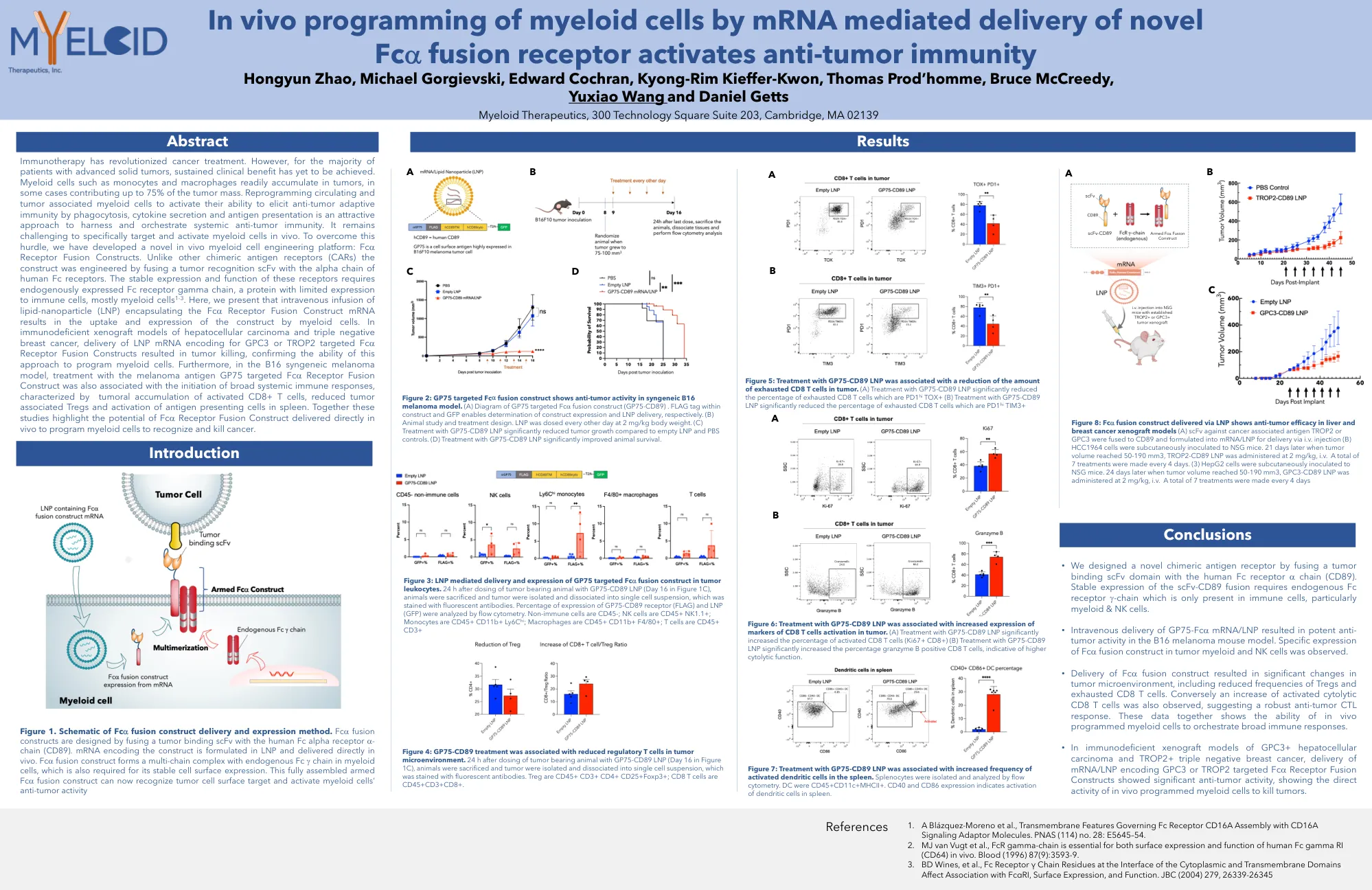
AACR2022体内递送海报
免疫疗法彻底改变了癌症治疗。但是,对于大多数晚期实体瘤患者,尚未实现持续的临床益处。髓样细胞(如单核细胞和巨噬细胞)很容易积聚在肿瘤中,在某些情况下,肿瘤质量的75%。重编程循环和肿瘤与髓样细胞相关,以激活其通过吞噬作用,细胞因子分泌和抗原表现来激活抗肿瘤适应性免疫的能力,是一种有吸引力的方法,可利用并策划系统性的抗肿瘤免疫。在体内专门靶向和激活髓样细胞仍然具有挑战性。为了克服这一障碍,我们开发了一种新型的体内髓细胞工程平台:FC A受体融合构建体。与其他嵌合抗原受体(CAR)不同,该构建体是通过将肿瘤识别SCFV与人体FC受体的α链融合而设计的。这些受体的稳定表达和功能需要内源表达的FC受体γ链,FC受体γ链是一种对免疫细胞表达有限的蛋白质,主要是髓样细胞1-3。在这里,我们介绍了包裹FC A受体融合构建体mRNA的静脉输注脂质 - 纳米颗粒(LNP)导致髓样细胞对构建体的摄取和表达。在肝细胞癌和三重阴性乳腺癌的免疫缺陷异种移植模型中,针对GPC3或trop2靶向FC的LNP mRNA的递送A受体融合构建体导致肿瘤杀死,从而确认了这种方法为骨髓细胞编程的能力。此外,在B16合成性黑色素瘤模型中,用黑色素瘤抗原GP75靶向FC A受体融合构建体的治疗也与启动广泛的全身免疫反应的启动有关,其特征在于肿瘤积累活化的CD8+ T细胞,可减少与肿瘤相关的TREG和SpleeNing spleen and spleen spleen and spleen的活化。这些研究共同强调了FC A受体融合构建体的潜力,直接在体内传递以编程髓样细胞以识别和杀死癌症。
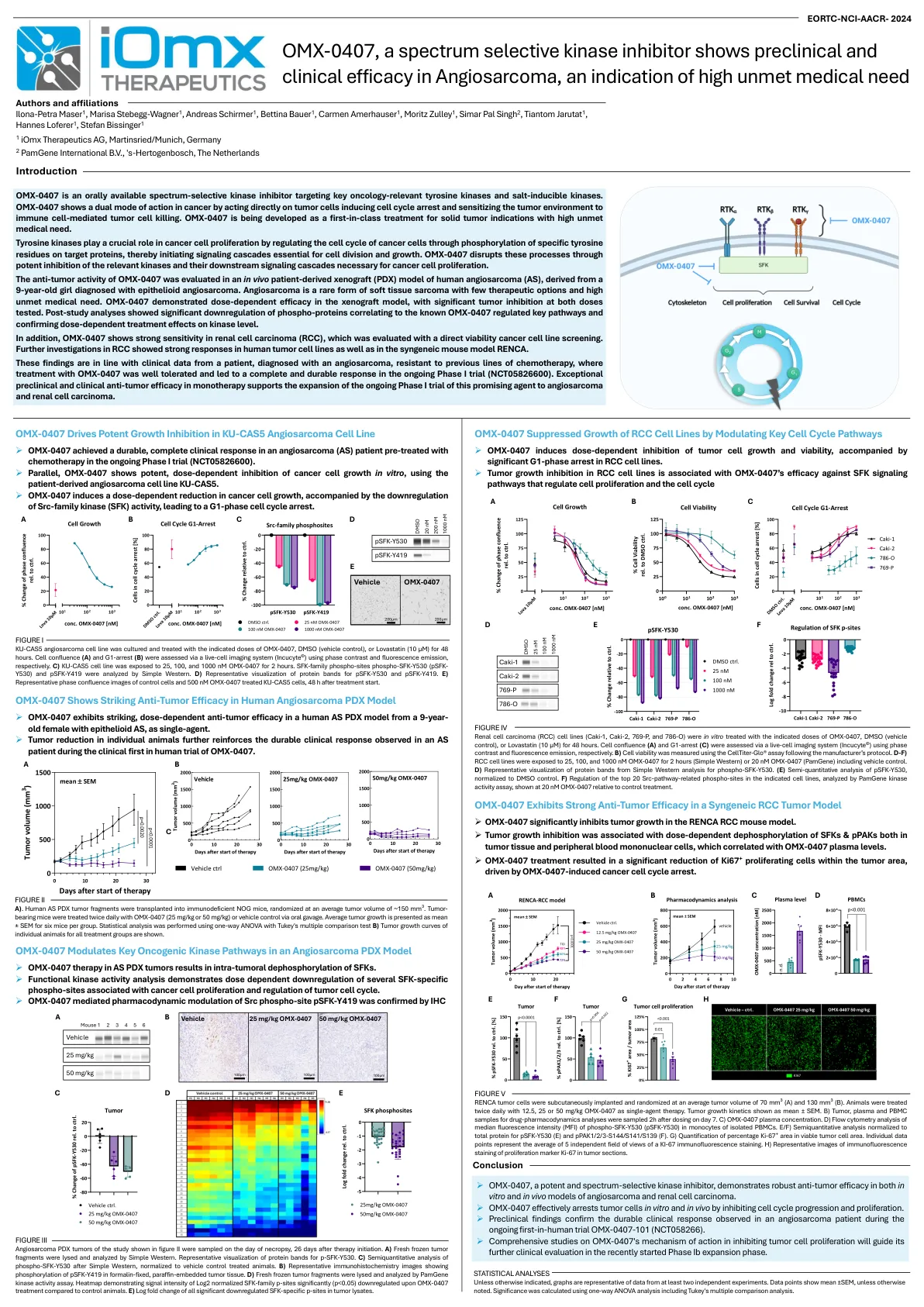
OMX-0407 是一种光谱选择性激酶抑制剂,表现出...
OMX-0407 是一种口服的光谱选择性激酶抑制剂,靶向关键的肿瘤相关酪氨酸激酶和盐诱导激酶。OMX-0407 在癌症中表现出双重作用模式,直接作用于肿瘤细胞,诱导细胞周期停滞,并使肿瘤环境对免疫细胞介导的肿瘤细胞杀伤敏感。OMX-0407 正在开发为针对具有高度未满足医疗需求的实体瘤适应症的同类首创治疗药物。酪氨酸激酶通过磷酸化靶蛋白上的特定酪氨酸残基来调节癌细胞的细胞周期,从而启动细胞分裂和生长所必需的信号级联,从而在癌细胞增殖中发挥关键作用。OMX-0407 通过有效抑制癌细胞增殖所必需的相关激酶及其下游信号级联来破坏这些过程。 OMX-0407 的抗肿瘤活性在人类血管肉瘤 (AS) 的体内患者异种移植 (PDX) 模型中得到评估,该模型来自一名被诊断为上皮样血管肉瘤的 9 岁女孩。血管肉瘤是一种罕见的软组织肉瘤,治疗选择很少,且未满足的医疗需求很高。OMX-0407 在异种移植模型中表现出剂量依赖性疗效,在两种测试剂量下均有显著的肿瘤抑制作用。研究后分析显示,与已知的 OMX-0407 调节关键通路相关的磷酸化蛋白显著下调,并证实了对激酶水平的剂量依赖性治疗效果。此外,OMX-0407 在肾细胞癌 (RCC) 中表现出很强的敏感性,这通过直接活力癌细胞系筛选进行了评估。对 RCC 的进一步研究表明,人类肿瘤细胞系以及同源小鼠模型 RENCA 均有强烈的反应。这些发现与一名被诊断患有血管肉瘤且对之前的化疗有抗药性的患者的临床数据相符,在正在进行的 I 期试验 (NCT05826600) 中,OMX-0407 的治疗耐受性良好,并产生了完全持久的缓解。单一疗法中出色的临床前和临床抗肿瘤效果支持将这种有前途的药物正在进行的 I 期试验扩展到血管肉瘤和肾细胞癌。

癌症免疫疗法学会(SITC)的共识定义,用于免疫检查点抑制剂相关的免疫相关事件(IRAES)术语
抽象背景黑色素瘤是一种免疫敏感疾病,如免疫检查点阻滞(ICB)的活性所证明,但许多患者将不反应或复发。最近,肿瘤浸润淋巴细胞(TIL)疗法已显示出ICB衰竭后黑色素瘤治疗的有希望的功效,表明细胞疗法的潜力。然而,由于转移了大量表型多样的T细胞,因此TIL处理伴随着制造局限性,产品异质性以及毒性问题。为了克服上述局限性,我们提出了一种受控的收养细胞治疗方法,其中T细胞由靶向SAR和黑色素瘤相关抗原的双特异性抗体(BIAB)选择性激活的合成激动受体(SAR)。方法是人类和鼠类SAR构建体的生成并转导到原代T细胞中。该方法在表达黑色素瘤相关抗原酪氨酸酶相关蛋白1(Tyrp1)和黑色素瘤相关软骨素硫酸盐蛋白聚糖(MCSP)(MCSP)(CSPG4)的鼠,人类和患者来源的癌症模型中得到了验证。sar t细胞。结果MCSP和Tyrp1表达在接受治疗和未经治疗的黑色素瘤的患者的样品中保守,支持其用作黑色素瘤靶标抗原。结论SAR T细胞 - 绝经方法提供了特定和条件T细胞激活在所有测试模型中,靶细胞和抗TYRP1×抗SAR或抗MCSP×抗SAR BIAB诱导的条件抗原依赖性激活,SAR T细胞的增殖和靶向肿瘤细胞裂解。在体内,抗肿瘤活性和长期生存是由SAR T细胞和BIAB在合成性肿瘤模型中的共同给药介导的,并在包括患者衍生的异种移植模型在内的几种异种移植模型中进一步验证。