XiaoMi-AI文件搜索系统
World File Search System变构

回顾一种姗姗来迟的针对非小细胞肺癌 KRAS 突变的靶向治疗:聚焦 Adagrasib
直到最近,尽管 KRAS 突变是最常见的致癌驱动因素之一,但其在实体瘤治疗中仍未得到满足。从历史上看,KRAS 突变很难靶向,这是因为其整体表面结构光滑,缺乏结合位点,尺寸小,且对 GTP/GDP 的亲和力极高。21、22 2013 年,Ostrem 等人首次发现了靶向 KRAS G12C 的潜力,他们发现结合变构口袋可以将 GDP 结合的 KRAS 锁定在其非活性状态。23 研究表明,RAS 蛋白第 12 或 13 个密码子的突变会损害 GTP 水解,使 RAS 处于 GTP 结合的活性状态。24 这导致了 sotorasib 的开发,这是同类中首批 KRASG12C 失活状态抑制剂之一,并最终促使 FDA 批准 sotorasib 用于治疗

引用本文:Chiara Milanese & Pier G. Mastroberardino (2020): DNA 损伤诱导的磷酸戊糖顺式通道增强作用的观点
在最近的一项研究中,我们描述了发生在小鼠模型和转录偶联和全球基因组核苷酸切除修复受损(分别为 TC-NER 和 GG-NER)患者标本中的代谢重排。在这里,我们描述了一种机制,将 DNA 修复缺陷导致的转录停滞与细胞内 ATP 水平增强联系起来,后者反过来变构抑制糖酵解酶 ATP 依赖性 6-磷酸果糖激酶(Pfk,最为人所知的是磷酸果糖激酶)通过戊糖磷酸途径(PPP)重新路由葡萄糖。PPP 的增强本质上与 NADPH 还原当量的产生增加有关——这些还原当量是在途径的氧化分支中产生的——在我们的实验系统中,氧化剂种类和/或内源性氧化还原酶活性的比例并不相符,因此最终导致还原应激 1(图 1A)。

大数据和人工智能发现针对无三维结构蛋白质的新型药物,并攻克无药可用靶点
摘要 靶向药物的发现很大程度上依赖于靶蛋白的三维结构,当未知蛋白质靶点的三维结构时,设计其对应的靶向药物非常困难。某些蛋白质(即所谓的不可成药靶点)尽管三维结构已知,但却缺乏针对它们的药物。随着蛋白质数据库中存储的晶体/低温电子显微镜结构越来越多,发现靶向药物的可能性大大增加。此外,通过识别先前不可成药的靶点的隐藏变构位点,也很有可能将之前的不可成药靶点转变为可成药靶点。本文主要介绍目前可用的针对未知三维结构的蛋白质发现新化合物的先进方法,以及如何将不可成药的靶点转变为可成药的靶点。

瞬时受体电位香草酸 1 在急性疼痛中的作用
TRPV1 在结构上被描述为同型四聚体通道。四个亚基中的每一个都含有六个跨膜结构域(S1-S6;图 2)。每个单体链总共由 838 个氨基酸组成,氨基酸残基 433–684 形成跨膜结构域。跨膜区由六个螺旋(S1-S6)组成,这些螺旋形成电压传感器样结构域(S1-S4)和内孔区(S5-S6)。跨膜结构域 5 和 6 由疏水 S4S5 连接环连接,并参与通道孔的形成。离子通道孔由选择性过滤器和孔螺旋形成。来自螺旋 S6 底部的残基充当激活门。不同的 TRPV 亚型具有不同的孔半径,可调节通道选择性。激活配体的结合导致两个门 8 的顺序和变构耦合打开。

药物靶标的结构研究和药物代谢酶
本文中介绍的工作描述了如何获取有关蛋白质的结构信息,以及如何使用它来回答有关蛋白质功能,动态行为以及与其他蛋白质或配体的相互作用的科学问题。嘧啶降解,药物代谢酶β-尿肽酶(βUP)的催化功能取决于寡聚态之间的变化。在二聚体二聚体界面中的氨基酸H173和H307在活性位点取代,表明这些对于βup激活至关重要。对基材和产物类似物的抑制作用研究允许假设与F205相互作用的能力可以将激活因子与抑制剂区分开。使用低温电子显微镜确定了激活的较高低聚物状态的第一个结构,并确认封闭的入口环构象和二聚体二聚体接口在HSβUP和DMβUP之间保守。研究了表观遗传药物靶标与含有蛋白3(SMYD3)的MYND结构域与可能的抑制剂之间的相互作用。晶体结构证实了在SMYD3的活性位点,合理设计的靶向抑制剂的共价键。使用具有阻塞活性位点的生物传感器屏幕发现了一个新的变构结合位点。晶体结构揭示了新结合位点的位置,以及变构抑制剂的(s) - (r)对映异构体的结合模式。最后,采用了一种基于碎片的药物发现方法,并通过碎片屏幕上的命中进行共结晶和浸泡Smyd3。这导致四个晶体结构在酶的几个位置的碎片密度弱。动态乙酰胆碱结合蛋白(ACHBP)是Cys环型配体离子通道的同源物。从各种生物传感器筛选中进行命中,其中一些表明构象变化与ACHBP共结晶。确定了来自生物物理筛选的命中化合物的复合物中ACHBP的七个晶体结构。在几个晶体结构中检测到Cys-loop的小构象变化,与生物传感器筛选的结果一致。在这些研究中,我们探讨了研究与药物发现和优化相关的蛋白质功能和调节的新策略。

通过化学神经解剖学和图像定量分析推断中枢神经系统中的细胞间通讯:对神经药理学的影响
摘要:在过去的几十年中,基于生化解剖学方法、显微镜和脑成像新技术以及所获图像的定量分析之间的协同作用,形态学研究获得了有关脑结构和功能的新证据。这一努力扩大了对脑结构的认识,将中枢神经系统描绘成一个巨大的细胞和区域网络,其中细胞间通讯过程不仅涉及神经元,还涉及其他细胞群,几乎决定了系统执行的整合功能的所有方面。本文描述了这些过程的主要特征。它们包括已确定的两种基本细胞间通讯模式(即布线和体积传输)以及调节细胞间信号传导的机制,例如共传递和变构受体-受体相互作用。这些特征也可能为开发治疗中枢神经系统疾病的新药理学方法开辟新的可能性。本文还将简要讨论这一方面,因为这可能对分子医学产生重大影响。

基于片段的共价配体发现
受最近批准用于癌症治疗的共价激酶抑制剂 (TKI) 的启发,共价探针和药物的开发经历了一场复兴,现在吸引了工业界和学术界的浓厚兴趣,包括针对 EGFR 的抑制剂:阿法替尼 (Gilotrif) 和奥希替尼 (Tagrisso) 或 BTK:阿卡替尼 (Calquence) 和伊布替尼 (Imbruvica)。1–4 与暂时靶向保守底物和/或变构结合位点的非共价小分子不同,共价抑制剂通常在效力、选择性、药代动力学和药效学方面表现出差异化的药理学,因为它们能够与靶蛋白形成不可逆的共价键。 5,6 尽管有这些优点,许多人仍然对共价抑制剂持怀疑态度,因为它们会产生能够引发特异性免疫反应和过敏/超敏反应的蛋白质加合物。7,8 从历史上看,共价药物的发现

有效和选择性Tyk2降解器,没有JAK活动,有效并完全抑制IL12/23和IFN-A信号通路
引言TYK2是激酶的JAK家族的成员,它结合了IL-12,IL-23和I型IFN受体,以募集和磷酸化信号转录器以及转录(STAT)转录因子的激活(STAT)。功能变异的丧失在自身免疫性疾病中具有保护性,而TYK2的变构抑制剂(Deucravacitinib)以及针对IL-12,IL-23和IFN-α的生物学剂已批准用于治疗多种自身免疫性疾病,使TYK2的治疗方法是具有极具吸引力的目标。tyk2,p1104a的常识变体,使蛋白质催化无效的蛋白质仍然支持通过I型IFN途径的信号传导,这表明抑制I型IFN需要阻止脚手架函数。此外,批准或临床发育中的TYK2抑制剂尚未在临床相关剂量时显示出完全靶标的抑制作用。
将Debio 0432识别为有效且有选择性的USP1 ...
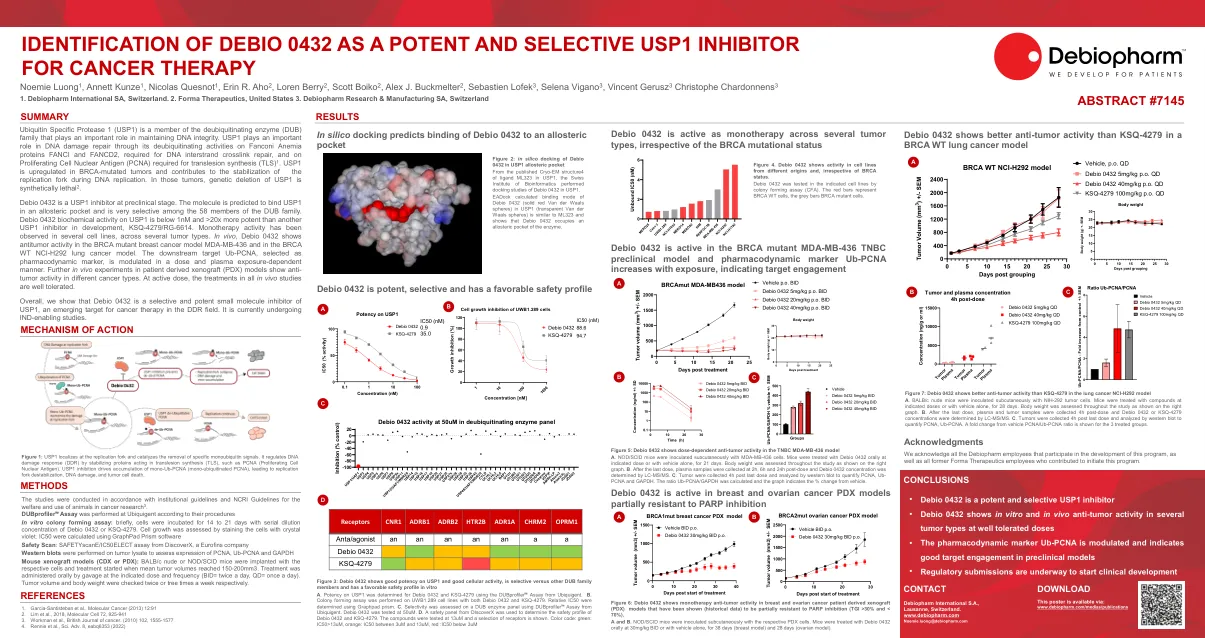
Debio 0432是临床前阶段的USP1抑制剂。该分子预计将在变构袋中结合USP1,并且在DUB家族的58个成员中非常有选择性。DEBIO 0432 USP1上的生化活性低于1NM低于1NM,比其他USP1抑制剂(KSQ-4279/RG-6614)高20倍。单一疗法活性。在体内,Debio 0432显示了BRCA突变乳腺癌模型MDA-MB-436和BRCA WT NCI-H292肺癌模型中的抗肿瘤活性。以剂量和血浆暴露依赖性方式调节下游目标UB-PCNA,被选为药效标记。进一步的患者衍生异种移植物(PDX)模型的体内实验显示了不同癌症类型的抗肿瘤活性。在活性剂量时,所有体内研究中的治疗方法都得到很好的耐受性。

2020-Small-TetR-FRET.pdf
在相关努力中,[10] 我们扩展了适用于均相 FRET 检测的分子识别元件列表,包括变构转录因子 (aTF),这是一类特定的底物结合蛋白,可在离散蛋白质结构域中结合 DNA 和小分子效应物。在这里,我们描述了使用特征明确的 aTF TetR 进行分子识别的其他新型传感器,使用改变 aTF-DNA 结合亲和力的 aTF 变体来调节传感器灵敏度,并展示了一种带有遗传编码供体荧光团的额外传感器设计。这些额外的传感器展示了我们方法的普遍性,同时详细介绍了一种更容易被各种研究小组采用的传感器设计。变构转录因子是调节蛋白,包含 DNA 结合结构域和效应物结合结构域,能够以高特异性和选择性识别小分子。 [11] 在目标分析物存在的情况下,aTF 对其 DNA 结合序列的亲和力会受到调节,从而促进下游基因表达的阻遏物或去阻遏物调节。[11] aTF 与其同源 DNA 和效应配体之间独特但相互关联的结合提供了一种内在的转导机制,我们将其与 FRET 偶联以进行光学读出。[10] 其他先前描述的基于底物结合蛋白的 FRET 传感器通过染料标记的配体的置换(竞争性测定)或蛋白质的构象变化来实现供体-受体距离的变化。[6,7] 我们的基于 aTF 的 FRET 传感器利用供体标记的 aTF 与其受体标记的同源 DNA 序列的分析物响应性解离来引起供体-受体距离的大幅变化。因此,这些 FRET 传感器无需对配体进行染料标记,因为染料标记会改变配体的结合行为 [12],同时能够通过供体和受体荧光团的完全解离产生显著的信号变化(图 1)。我们之所以选择 TetR 进行这项研究,是因为它是一种特性良好的 aTF,在实验室环境中广泛用于基因调控和诱导蛋白表达。[11] TetR