XiaoMi-AI文件搜索系统
World File Search System强效

产品专论包括患者...
• pralsetinib 与中 – 强效 CYP3A4 和/或 P-gp 抑制剂共同给药可能会增加 pralsetinib 的血浆浓度,从而增加不良反应的风险。避免将 GAVRETO 与中 – 强效 CYP3A4 和/或 P-gp 抑制剂共同给药。如果无法避免共同给药,请降低 GAVRETO 剂量(见 4.2 推荐剂量和剂量调整和 9 种药物相互作用)。 • pralsetinib 与强效或中效 CYP3A4 诱导剂共同给药可能会降低 pralsetinib 的血浆浓度,并可能导致 pralsetinib 的疗效降低。避免将 GAVRETO 与强效或中效 CYP3A4 诱导剂共同给药。如果无法避免共同给药,请增加 GAVRETO 剂量(见 4.2 推荐剂量和剂量调整和 9 种药物相互作用)。

Rinvoq, INN-Upadacitinib
当与强效 CYP3A4 抑制剂(如酮康唑、伊曲康唑、泊沙康唑、伏立康唑、克拉霉素和葡萄柚)共同使用时,Upadacitinib 的暴露量会增加。在一项临床研究中,乌帕替尼与酮康唑共同给药导致乌帕替尼 Cmax 增加 70%,AUC 增加 75%。对于长期接受强效 CYP3A4 抑制剂治疗的患者,应谨慎使用每日一次 15 毫克的乌帕替尼。对于长期接受强效 CYP3A4 抑制剂治疗的特应性皮炎患者,不建议使用每日一次 30 毫克的乌帕替尼。对于服用强效 CYP3A4 抑制剂的溃疡性结肠炎患者,建议起始剂量为每天一次 30 毫克(最多 16 周),建议维持剂量为每天一次 15 毫克(见 4.2 节)。对于长期使用,应考虑强效 CYP3A4 抑制剂的替代品。在使用乌帕替尼治疗期间应避免食用含有葡萄柚的食物和饮料。

混合免疫产生的抗 S1 和抗 S2 抗体可引发针对 SARS-CoV-2 的强效交叉变异 ADCC
简介感染和接种全球使用的任何一种主要 COVID-19 疫苗均可诱导针对 SARS-CoV-2 刺突 (S) 蛋白的体液免疫,其中大多数疫苗将 S 编码为单一抗原 (1–3)。抗 S 抗体靶向蛋白质内的多个区域,但主要关注的是中和无细胞病毒体的区域。这些抗体主要结合在受体结合结构域 (RBD) 内,在某些情况下结合在 N 端结构域 (NTD) 内,这两个结构域均位于蛋白质的 S1 结构域中。中和抗体可阻断或阻止 SARS-CoV-2 与进入受体血管紧张素转换酶 2 (ACE-2) 之间的结合,或阻止病毒进入所需的结合后事件 (4, 5)。它们被认为对于减少 SARS-CoV-2 的传播至关重要;因此,它们是预测 COVID-19 疫苗效力的关键指标 (6)。尽管中和抗体的重要性显而易见,但它们也有公认的局限性。中和表位的数量有限,导致 SARS-CoV-2 变体被快速选择,这些变体的突变会削弱抗体与关键中和位点的结合 (7, 8)。在人类群体中进化了大约 3 年后,令人担忧的 SARS-CoV-2 变体已基本摆脱了由祖先 S 抗原诱导的抗体的中和活性,并不断进化以逃避由较新的变体感染诱导的抗体。因此,疫苗在接种后的数月内,其预防感染的效力已经降低。一旦发生感染,SARS-CoV-2 可以直接在细胞间传播,进一步削弱中和抗体的效力 (9)。为了抵消细胞间病毒传播,抗体需要识别受感染细胞表面的病毒抗原,而不是中和无细胞病毒体 (10)。这些抗体会招募效应细胞(如 NK 细胞)来

通过基于工程化套索肽的强效和选择性双重 αvβ6/8 抑制剂克服免疫检查点抑制剂耐药性
肿瘤微环境 (TME) 中的整合素 v 6 和 v 8 已被证实能激活免疫抑制 TGF- ,这是一系列肿瘤对免疫检查点抑制剂产生耐药性的重要机制。在本研究中,我们展示了套索肽作为设计新疗法的多功能支架的效用。通过结合表位扫描、计算设计和定向进化,设计了一系列高效且选择性的双重 v 6/8 抑制剂。几种类似物,如套索肽 36 和 47 ,已被充分表征,并报告了物理化学、体外药理学和体内数据。套索肽 47 是 36 的一种半衰期延长衍生物,与检查点抑制剂联合使用时,已被证实可强烈增强小鼠抗 mPD-1 耐药肿瘤的敏感性。研究表明,47/抗 mPD-1 组合可在三阴性乳腺癌和卵巢癌小鼠模型中阻止肿瘤生长并使肿瘤消退。因此,TME 中表达的 v 6/8 整合素的双重抑制代表了一种有前途的肿瘤特异性策略,可克服 TGF- 驱动的耐药性并增强免疫检查点抑制剂的抗肿瘤功效。_________________________________________________

新型功能性抗 HER3 单克隆抗体,对多种人类上皮癌具有强效抗癌作用
进行性癌症对化疗的耐药性是一个严重的临床问题。在这种情况下,人表皮生长因子受体 3 (HER3) 在 HER1 和 HER2 靶向治疗的耐药性中起着重要作用。由于抗 HER3 单克隆抗体 (mAb)(例如 patritumab)的临床试验与现有药物相比未能显示出显著的效果,因此我们生成了针对抗 HER3 的新型 mAb。新型大鼠 mAb 与表达 HER3 的 HEK293 细胞发生反应,但不与表达 HER1、HER2 或 HER4 的细胞发生反应。siRNA 敲低和基于 CRISPR/Cas9 的基因组编辑敲除导致 mAb 结合丧失,从而证实了 mAb 的特异性。CDR 序列和种系片段的分析显示,七种 mAb 分为四组,而 patritumab 的结合被七种 mAb 中的一种抑制。七种 mAb 已显示出与各种人类上皮癌细胞的反应性、细胞表面 HER3 的强内化活性以及对 NRG1 结合、NRG1 依赖性 HER3 磷酸化和细胞生长的抑制作用。抗 HER3 mAb 还与体内肿瘤组织和癌症组织来源的球体反应。Ab4 抑制了裸鼠体内人类结肠癌细胞的肿瘤生长。目前的 mAb 可能优于现有的抗 HER3 mAb,并支持现有的抗癌治疗性 mAb。

利用基于白蛋白结合域衍生亲和蛋白的强效药物偶联物靶向 HER2 表达肿瘤
摘要:白蛋白结合域衍生的亲和蛋白 (ADAPT) 是一类小型折叠工程支架蛋白,在靶向癌症肿瘤方面具有巨大前景。在这里,我们通过与白蛋白结合域 (ABD) 融合延长了 ADAPT 的体内半衰期,该蛋白靶向人表皮生长因子受体 2 (HER2),并用高细胞毒性有效载荷 mertansine (DM1) 武装它,以研究其体外和体内特性。所得药物偶联物 ADAPT6-ABD-mcDM1 保留了与其预期靶标(即 HER2 和血清白蛋白)的结合。此外,它能够特异性地结合具有高 HER2 表达的细胞,被内化,并显示出强毒性,IC 50 值范围为 5 至 80 nM。相反,对于具有低 HER2 表达的细胞没有发现毒性作用。体内实验中,用 99m Tc 放射性标记的 ADAPT6-ABD-mcDM1 在大多数正常器官中的摄取率较低,主要排泄途径为肾脏。24 小时后肿瘤的摄取率为 5.5% ID/g,高于除肾脏外所有正常器官在此时间点的摄取率。通过预先注射过量的单克隆抗体曲妥珠单抗(在 HER2 受体上具有重叠表位)可阻断肿瘤的摄取。总之,基于亲和蛋白 ADAPT 平台的半衰期延长药物偶联物有望进一步发展为靶向癌症治疗。

![清肠和免疫补充剂评论(最佳客户结果)副作用、成分 [3RQNP]](/simg/3\3f800fe4b897c71c3db4030700c401fc038d9cd2.webp)

混合免疫产生的抗 S1 和抗 S2 抗体可引发针对 SARS-CoV-2 的强效交叉变异 ADCC
简介感染和接种全球使用的任何一种主要 COVID-19 疫苗均可诱导针对 SARS-CoV-2 刺突 (S) 蛋白的体液免疫,其中大多数疫苗将 S 编码为单一抗原 (1–3)。抗 S 抗体靶向蛋白质内的多个区域,但主要关注的是中和无细胞病毒体的区域。这些抗体主要结合在受体结合结构域 (RBD) 内,在某些情况下结合在 N 端结构域 (NTD) 内,这两个结构域均位于蛋白质的 S1 结构域中。中和抗体可阻断或阻止 SARS-CoV-2 与进入受体血管紧张素转换酶 2 (ACE-2) 之间的结合,或阻止病毒进入所需的结合后事件 (4, 5)。它们被认为对于减少 SARS-CoV-2 的传播至关重要;因此,它们是预测 COVID-19 疫苗效力的关键指标 (6)。尽管中和抗体的重要性显而易见,但它们也有公认的局限性。中和表位的数量有限,导致 SARS-CoV-2 变体被快速选择,这些变体的突变会削弱抗体与关键中和位点的结合 (7, 8)。在人类群体中进化了大约 3 年后,令人担忧的 SARS-CoV-2 变体已基本摆脱了由祖先 S 抗原诱导的抗体的中和活性,并不断进化以逃避由较新的变体感染诱导的抗体。因此,疫苗在接种后的数月内,其预防感染的效力已经降低。一旦发生感染,SARS-CoV-2 可以直接在细胞间传播,进一步削弱中和抗体的效力 (9)。为了抵消细胞间病毒传播,抗体需要识别受感染细胞表面的病毒抗原,而不是中和无细胞病毒体 (10)。这些抗体会招募效应细胞(如 NK 细胞)来
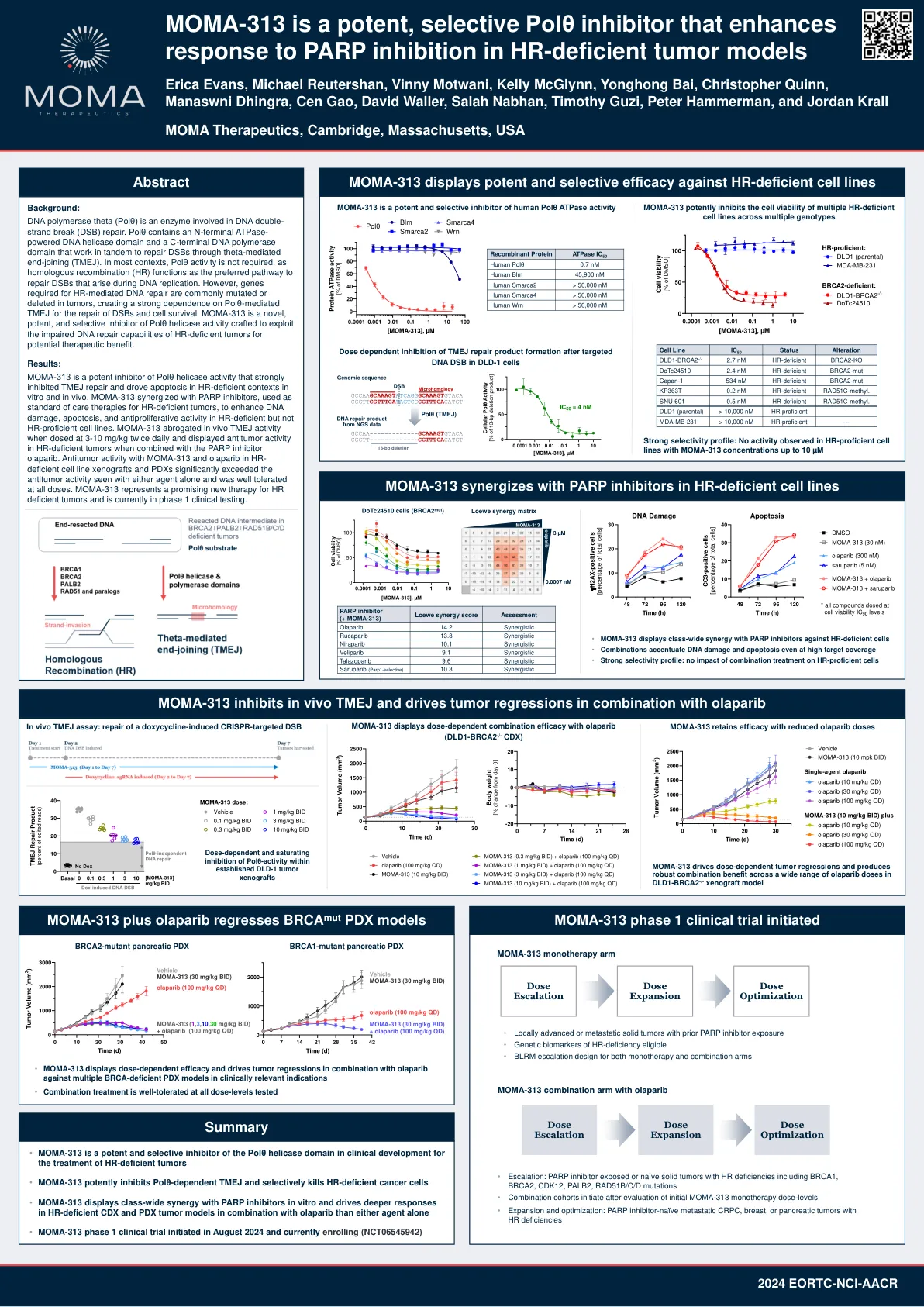
MOMA-313 是一种强效、选择性 Polθ 抑制剂,可增强 HR 缺陷肿瘤模型对 PARP 抑制的反应
DNA 聚合酶 theta (Polθ) 是一种参与 DNA 双链断裂 (DSB) 修复的酶。Polθ 包含一个 N 端 ATPase 驱动的 DNA 解旋酶结构域和一个 C 端 DNA 聚合酶结构域,它们协同作用,通过 theta 介导的末端连接 (TMEJ) 修复 DSB。在大多数情况下,Polθ 活性不是必需的,因为同源重组 (HR) 是修复 DNA 复制过程中出现的 DSB 的首选途径。然而,HR 介导的 DNA 修复所需的基因通常在肿瘤中发生突变或缺失,导致 DSB 修复和细胞存活严重依赖 Polθ 介导的 TMEJ。MOMA-313 是一种新型、有效且选择性的 Polθ 解旋酶活性抑制剂,旨在利用 HR 缺陷型肿瘤受损的 DNA 修复能力来获得潜在的治疗益处。