XiaoMi-AI文件搜索系统
World File Search System抗增殖

二叶林的抗增殖和抗迁移活性...
癌症是每年影响数百万人的重大健康负担。结直肠癌 (CRC) 是第三大常见恶性肿瘤,而且不再仅限于发达国家。1 一般来说,癌症治疗包括手术、化疗和放疗。治疗策略的选择取决于肿瘤在原发组织和转移组织中的扩散情况,通常,联合治疗更受青睐。2 虽然细胞毒性化疗是消除呈指数级分裂的恶性细胞的主要选择,但它会产生不良反应。3 已经引入了更具体的抗癌靶向疗法,以最大限度地减少对正常细胞的毒性。尽管如此,靶向疗法通常与细胞毒性化疗相结合,以获得更好的临床效果。传统化疗的另一个主要问题是产生耐药性,而通过在多个层面靶向肿瘤细胞可能会取代这种耐药性。这些耐药细胞在治疗中存活下来并迁移到远处器官产生转移。4 旨在诱导细胞凋亡并抑制增殖、迁移和血管生成的理想抗癌药物可以提供良好的临床效果。现有的细胞毒性和靶向治疗药物可杀死快速增殖的细胞或靶向参与转移和血管生成的特定蛋白质。模式已转变为开发具有多方面作用和独特机制的抗癌药物。二叶酸是一种天然存在于许多植物物种中的糖苷,化学上与芳基萘配体有关。二叶酸的药理作用主要归因于其与

细辛醚在乳腺癌细胞系中具有抗增殖潜力
乳腺癌是全球面临的重大健康挑战,需要不断探索新的治疗方法。细辛酮是一种来自菖蒲属的生物活性化合物,具有良好的抗癌特性,但其对乳腺癌细胞的影响尚未得到充分研究。本研究使用体外和计算机模拟方法研究了细辛酮对乳腺癌细胞系的抗癌潜力。使用 DPPH 自由基清除试验评估了细辛酮的抗氧化活性,结果显示其对自由基具有剂量依赖性(25.56%、32.18%、47.73%、54.83% 和 66.74%)。MTT 试验显示细胞活力呈剂量依赖性下降,表明细辛酮对乳腺癌细胞具有细胞毒性。 mRNA 表达分析表明,针对凋亡调节因子,例如 Bax(1、1.3、1.52 倍变化上调)和 Bad(1、1.4 和 1.6 倍变化上调)基因表达,表明细辛醚通过内在途径诱导细胞凋亡。此外,细辛醚抑制 Akt mNRA(1、0.6 和 0.4 倍变化下调)、caspase-3(1、1.4 和 1.7 倍变化上调)和细胞色素 c mRNA(1、1.2 和 1,54 倍变化上调),表明干扰关键的癌症进展途径。分子对接研究预测细辛醚与参与细胞凋亡和细胞存活的关键蛋白质(包括 Bax、Bad、细胞色素 c、caspase 3 和 Akt)之间存在有利的结合相互作用。这些发现共同强调了细辛醚对乳腺癌细胞的多方面抗癌机制。这项研究强调了阿魏酸作为乳腺癌天然治疗剂的潜力,为进一步探索转化研究和临床试验提供了途径。本研究大大提高了我们对阿魏酸抗癌特性的认识,为开发新的有效乳腺癌疗法提供了有希望的方向。

EGFR 抑制剂西妥昔单抗的抗增殖作用和...
在癌症免疫治疗方法中,最常转移和批准用于临床治疗的是单克隆抗体(7)。西妥昔单抗是目前作为药物生产的主要抗体之一。西妥昔单抗是一种靶向 EGFR 的单克隆抗体,临床批准用于癌症免疫治疗。西妥昔单抗是一种嵌合抗体,这意味着它同时包含人类和小鼠蛋白质序列(8)。表皮生长因子受体 (EGFR) 是一种跨膜糖蛋白。它是 I 型受体酪氨酸激酶亚家族的成员,包括 HER1、HER2、HER3 和 HER4。EGFR 在大多数正常上皮组织中组成性表达(9)。已确定 EGFR 在许多癌症中过表达。EGFR 过表达与预后不良、总生存期缩短和/或转移风险增加有关。蛋白酪氨酸激酶的活性受到严格调控,因为它们是细胞生长、分化和死亡的介质 (10)。EGFR 抑制剂用于治疗不同类型的癌症,在这些癌症中,已发现 RTK 家族失调,从而导致

RHOB在糖皮质激素受体对巨噬细胞RAW264.7细胞的抗增殖作用中的作用
尽管据报道糖皮质激素(GC)抑制巨噬细胞杀伤性和甲状腺因子产生对促炎的刺激,但GC对巨噬细胞增殖的影响是有争议的。在我们先前的研究中,我们发现在鼠巨噬细胞系RAW264.7细胞(RAW-Gr(k)细胞)中抑制糖皮质激素受体(GR)的表达,可显着促进细胞增殖。在本研究中,我们提供了一个证据,表明RhoB的表达是具有抗癌特征的Rho GTPase的成员,在RAW-GR(K)和RAW264.7细胞中急剧降低了用GR-RNAI载体转染。在RAW -gr(K)和RAW264.7CELLSBYBYTRANSFETECTION WITHWILD -WILD -TYPE RHOB表达载体(RHOB -WT)或组成激活的RHOB质粒(RHOB -V14)中,RHOB的过表达或本构激活导致两种细胞系的增殖降低。相反,RAW264.7细胞的增殖为

吲哚或2-氧烷基杂种的主要生物目标作用是有希望的抗增殖剂
吲哚部分被认为是一种独特的核心支架,可以与不同类型的基因和蛋白质结合,并且具有易于合成技术和独家化学特性。这些特征使基于吲哚的支架成为药物化学研究化学家的主要探测器。利用吲哚部分的杂交技术可以提高功效,打击耐药性并降低最终化合物的副作用。 因此,最近已经报道了许多基于吲哚和2-氧气吲哚的杂种,并进行了临床前和临床研究。 但是,除了在不久的将来开发更有效的基于吲哚的脚手座,还可以在多静脉药药物疗法中获得更多的成就,但仍有更多的研究工作对于清楚地了解癌症治疗中的癌症起源和耐药性机制至关重要。 在这项综述研究中引入的这些吲哚和基于2-氧烷基的杂种的有前途的抗增生活性背后,有四种主要机制是蛋白激酶,DNA拓扑异构酶,组蛋白脱乙酰基酶(HDAC)和tubulin聚合抑制活性。 在此,这篇综述将简要说明新合成的吲哚和2-氧气吲哚的混合动力及其多种机制,以展示其有希望的抗增生活性,这将是进一步改善药物发明和消除耐药性问题的方法的宝贵步骤。利用吲哚部分的杂交技术可以提高功效,打击耐药性并降低最终化合物的副作用。因此,最近已经报道了许多基于吲哚和2-氧气吲哚的杂种,并进行了临床前和临床研究。但是,除了在不久的将来开发更有效的基于吲哚的脚手座,还可以在多静脉药药物疗法中获得更多的成就,但仍有更多的研究工作对于清楚地了解癌症治疗中的癌症起源和耐药性机制至关重要。在这项综述研究中引入的这些吲哚和基于2-氧烷基的杂种的有前途的抗增生活性背后,有四种主要机制是蛋白激酶,DNA拓扑异构酶,组蛋白脱乙酰基酶(HDAC)和tubulin聚合抑制活性。在此,这篇综述将简要说明新合成的吲哚和2-氧气吲哚的混合动力及其多种机制,以展示其有希望的抗增生活性,这将是进一步改善药物发明和消除耐药性问题的方法的宝贵步骤。
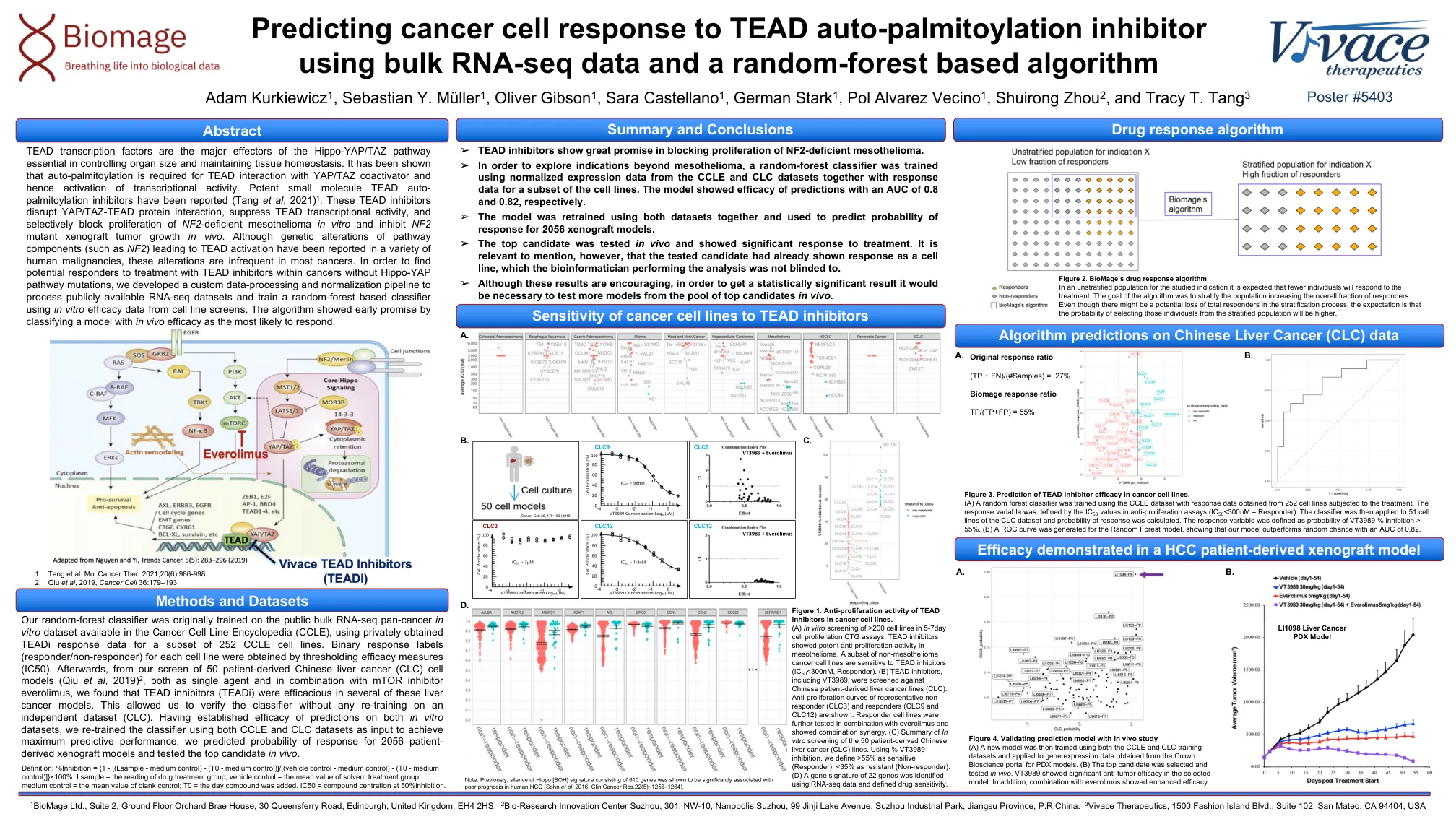
预测癌细胞对TEAD自动甲虫素的反应...
图1。癌细胞系中TEAD抑制剂的抗增殖活性。 (a)5-7天细胞增殖CTG分析中> 200个细胞系的体外筛选。 块状抑制剂在间皮瘤中显示出有效的抗增殖活性。 非间皮瘤癌细胞系的子集对TEAD抑制剂(IC 50 <300nm,响应者)敏感。 (b)在包括中国患者衍生的肝癌系(CLC)的情况下,筛选了包括VT3989在内的TEAD抑制剂。 显示了代表性非响应者(CLC3)和响应者(CLC9和CLC12)的抗增殖曲线。 响应者细胞系与依维莫司(Everolimus)结合进一步测试,并显示出协同作用。 (c)50例患者介绍的中国肝癌(CLC)系的体外筛查摘要。 使用%VT3989抑制,我们将> 55%定义为敏感(响应者); <35%的抗性(无反应器)。 (d)使用RNA-seq数据和定义的药物敏感性鉴定了22个基因的基因特征。癌细胞系中TEAD抑制剂的抗增殖活性。(a)5-7天细胞增殖CTG分析中> 200个细胞系的体外筛选。块状抑制剂在间皮瘤中显示出有效的抗增殖活性。非间皮瘤癌细胞系的子集对TEAD抑制剂(IC 50 <300nm,响应者)敏感。(b)在包括中国患者衍生的肝癌系(CLC)的情况下,筛选了包括VT3989在内的TEAD抑制剂。显示了代表性非响应者(CLC3)和响应者(CLC9和CLC12)的抗增殖曲线。响应者细胞系与依维莫司(Everolimus)结合进一步测试,并显示出协同作用。(c)50例患者介绍的中国肝癌(CLC)系的体外筛查摘要。使用%VT3989抑制,我们将> 55%定义为敏感(响应者); <35%的抗性(无反应器)。(d)使用RNA-seq数据和定义的药物敏感性鉴定了22个基因的基因特征。

DNA修复途径是与曲妥珠单抗Deruxtecan在HER2靶向的抗体抗体 - 毒物结合物中耐药的耐药性HER2过表达
蛋白质还原似乎在我们观察到HER2指导的ADC耐药细胞系中HER2表达降低的过程中起作用。确认降低HER2蛋白水平是否是对T-DXD抗性的原因,我们在T-DXD耐药的BC细胞系中过表达HER2,并测量了T-DXD的抗增殖作用。HER2的过表达不会诱导T-DXD的抗增殖作用(sup。 图4A)。 表明,HER2定向ADC抗性细胞系中HER2的表达降低足以进行ADCHER2的过表达不会诱导T-DXD的抗增殖作用(sup。图4A)。 表明,HER2定向ADC抗性细胞系中HER2的表达降低足以进行ADC图4A)。表明,HER2定向ADC抗性细胞系中HER2的表达降低足以进行ADC

2025; 21(4): 1545-1565. doi: 10.7150/ijbs.102079 研究论文高剂量抗坏血酸表现出抗增殖和抗侵袭作用,取决于
子宫内膜癌 (EC) 是最常见的妇科恶性肿瘤,通常以 PTEN 缺失、AKT/mTOR 通路激活为特征,对于复发和晚期患者有效治疗选择有限。高剂量抗坏血酸或与其他化疗药物联合使用在体内和体外均显示出强大的抗肿瘤作用。在本研究中,高剂量抗坏血酸显着抑制细胞增殖和侵袭,增加细胞应激和 DNA 损伤,并诱导 EC 细胞的细胞周期停滞和细胞凋亡。口服或腹膜内注射高剂量抗坏血酸 4 周可有效抑制 LKB1 fl/fl p53 fl/fl 小鼠 EC 模型中的肿瘤生长,且腹膜内注射比口服更有效。N-乙酰半胱氨酸部分逆转了抗坏血酸在 EC 细胞中的抗肿瘤作用和 LKB1 fl/fl p53 fl/fl 小鼠的肿瘤生长。通过 shRNA 敲低 PTEN 降低了 EC 细胞对抗坏血酸的抗肿瘤敏感性,而通过 Ipatasertib 抑制 AKT/mTOR 通路则显著增强了抗坏血酸在 EC 细胞中的抗肿瘤活性。与单独使用任一药物相比,抗坏血酸与紫杉醇联合使用可协同抑制 LKB1 fl/fl p53 fl/fl 小鼠中的肿瘤生长。总体而言,高剂量抗坏血酸部分通过 PTEN/AKT/mTOR 和细胞应激通路表现出抗肿瘤活性,并且在 EC 中与紫杉醇联合使用时这些抗肿瘤作用增强。抗坏血酸与紫杉醇联合使用的临床试验值得在 EC 患者中进一步研究。

文章新型抗增殖药物联苯烟酰胺:与顺铂和长春花碱相比,其对 MCF-7 细胞作用的 NMR 代谢组学研究
摘要:对用新型烟酰胺衍生物 (DT-8) 处理的 MCF-7 细胞系进行了基于 1 H-NMR 的代谢组学研究,并与两种具有明确作用机制的药物进行了比较,即 DNA 金属化药物顺铂 (顺式二氨二氯铂 (II),CDDP) 和抗有丝分裂药物长春花碱 (长春花碱,VIN)。通过细胞裂解物的 1 H-NMR 和光谱数据的多变量分析 (MVA),研究了这三种化合物(每种化合物的浓度对应于 IC 50 值)相对于对照组 (K) 的影响。发现不同治疗组的代谢特征与对照组存在相关差异。DT-8 与 K 和 VIN 与 K 的代谢特征有很大的重叠,表明生物反应和作用机制相似,与 CDDP 相比有显著差异。另一方面,DT8 似乎通过一种暗示蛋氨酸耗竭和/或 S-腺苷甲硫氨酸 (SAM) 限制的机制,扰乱有丝分裂纺锤体并最终阻止细胞分裂。
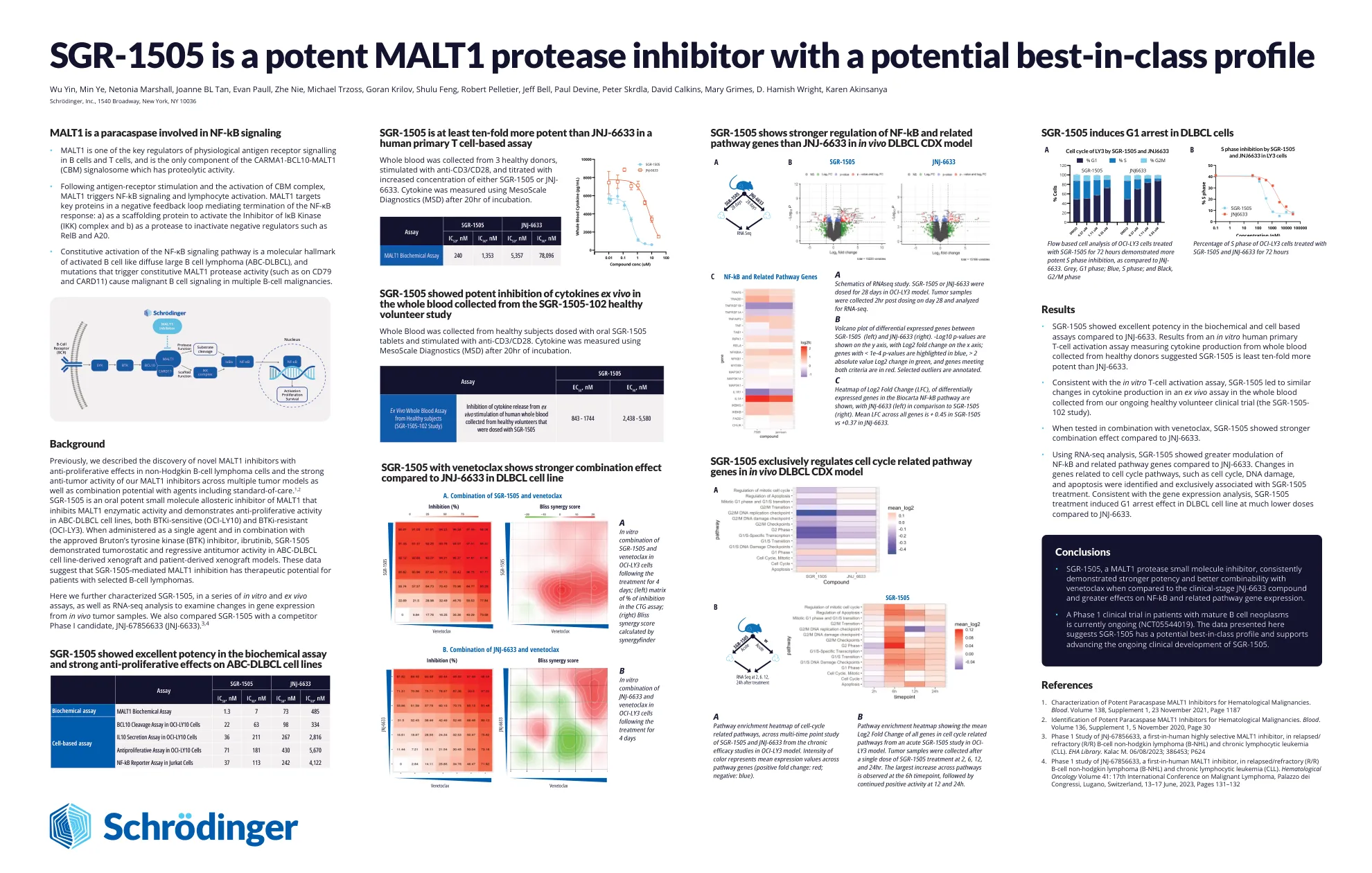
SGR-1505是一种有效的MALT1蛋白酶抑制剂,具有潜在的一流概况
先前,我们描述了在非霍奇金B细胞淋巴瘤细胞中具有抗增殖作用的新型MALT1抑制剂的发现,以及在多个肿瘤模型中MALT1抑制剂的强抗肿瘤活性,以及与包括标准护理在内的药物的组合。1,2 SGR-1505是MALT1的口服有效的小分子变构抑制剂,可抑制MALT1酶活性,并在ABC-DLBCL细胞系中表现出抗增殖活性,均为BTKI敏感性(OCI-LY10)(OCI-LY10)和BTKI-RESIS耐药(OCI-Ly3)。当用作单一药物并与批准的布鲁顿酪氨酸激酶(BTK)抑制剂相结合时,ibrutinib ibrutinib sgr-1505显示出在ABC-DLBCL细胞线衍生的Xenograft中表现出肿瘤抑制和回归抗肿瘤活性。这些数据表明,SGR-1505介导的MALT1抑制作用对于选定的B细胞淋巴瘤患者具有治疗潜力。