XiaoMi-AI文件搜索系统
World File Search System肌萎缩


腓骨肌萎缩症治疗进展
腓骨肌萎缩症 (CMT) 是一种遗传性神经肌肉疾病,可导致运动/感觉神经病变和代谢紊乱。它是最常见的遗传性运动和感觉神经病变,因此对于初级保健医生来说,这是一种需要密切关注的重要疾病。1 根据遗传学和表现,该疾病有多种不同的类型。CMT 1 型是由周围神经脱髓鞘引起的。1A 型是由于 PMP22 基因突变引起的。1B 型是由于 MPZ 基因突变引起的。脱髓鞘会导致传导速度降低,从而导致虚弱和麻木。CMT 2 型是由于轴突变性而不是脱髓鞘引起的。2A 型是由于 MFN2 突变引起的。最后,CMT X 型和 4 型也是脱髓鞘性神经病变。X 型是由于 PRPS1 基因突变引起的,而 4 型是由于 SH3TC2 的常染色体隐性突变引起的。 2 初级保健提供者需要注意三种不同的 CMT 表现。第一种是 10-30 岁的患者,双腿无力和感觉丧失的隐匿性、进行性发作,最终发展到手部。第二种是年幼的儿童,行走延迟、笨拙和跌倒。第三种是 40 岁以后发作的患者,有类似的体征和症状。1 目前,CMT 没有医疗治疗选择。唯一的选择是针对虚弱的综合康复计划,以及针对任何畸形的可能矫形手术或矫形器。锻炼是另一个重要方面,因为患者应该定期进行低强度锻炼。3 由于 CMT 是一种遗传性疾病,因此有很多机会可以针对不同类型的基因产物进行治疗。为了帮助医生,我重点介绍了目前正在研究的主要产品。

肌萎缩侧索硬化症的最新进展
摘要 近几年来,我们对 ALS 疾病分子机制的理解取得了长足进步,并迈出了将新研究成果(包括基因治疗方法)转化为临床实践的第一步。同样,在日益复杂的多学科行动背景下,辅助技术的最新出现也大大提高了采用更加个性化的支持和对症治疗方法的可能性,而这仍然是 ALS 管理的基石。在这种快速发展的背景下,我们在此全面介绍了有助于我们了解 ALS 发病机制的最新研究、临床试验的最新结果以及改善 ALS 患者临床管理的未来方向。

脊髓性肌萎缩症的药物治疗
▪ 1 型 SMA 是最常见的类型,影响 6 个月以下的婴儿。患者会感到严重虚弱,无法独自坐立。他们还可能出现呼吸困难和吞咽困难,头部控制能力差。如果不进行治疗,许多患者可能因呼吸衰竭而无法活过 2 岁。

肌萎缩症中的生物医学信号和机器学习......
摘要简介:机器学习 (ML) 技术在医疗保健领域的应用包含一个新兴概念,该概念设想为治疗罕见疾病做出巨大贡献。在这种情况下,肌萎缩侧索硬化症 (ALS) 涉及尚未揭开神秘面纱的复杂性。在 ALS 中,生物医学信号表现为潜在的生物标记,当与智能算法结合使用时,可用于疾病背景下的应用。方法:本系统文献综述 (SLR) 包括搜索和调查使用与 ALS 相关的 ML 技术和生物医学信号的原始研究。根据 SLR 协议的定义和执行,18 篇文章符合纳入、排除和质量评估标准,并回答了 SLR 研究问题。讨论:根据结果,我们确定了 ALS 背景下的三类与生物医学信号相结合的 ML 应用:诊断(72.22%)、通信(22.22%)和生存预测(5.56%)。结论:已报告了不同的算法模型和生物医学信号,并提出了有前途的方法,无论其类别如何。总之,本 SLR 概述了所分析的主要研究以及在 ALS 范围内构建和发展基于技术的研究的方向。

肌萎缩侧索硬化症 (ALS) 8 型
肌萎缩侧索硬化症 8 型 (ALS8) 是一种罕见的家族性 ALS 亚型,由囊泡相关膜蛋白相关蛋白 B (VAPB) 基因突变引起,特别是 p.P56S 突变。与散发性 ALS 相比,该病的特点是病情进展较慢、发病较早,并具有独特的临床特征,例如严重痉挛、肌束震颤、姿势性震颤以及认知和行为障碍。尽管目前的药物选择(例如利鲁唑、依达拉奉和苯丁酸钠/牛磺熊二醇)提供了适度的益处,但它们未能解决 ALS8 的潜在遗传机制。新兴的基因疗法、基于 RNA 的干预措施和干细胞方法有望实现精准治疗,但在临床应用中面临挑战。症状管理策略(包括呼吸、营养和心理支持)对于改善患者预后和生活质量至关重要。尽管在了解 ALS8 的遗传和分子发病机制方面取得了重大进展,但其稀有性、表型多变性和有限的临床数据对治疗进展构成了挑战。本篇叙述性综述重点介绍了当前的治疗策略、ALS8 的独特临床轨迹以及创新、亚型特异性干预的潜在途径,强调需要采取多学科和有针对性的方法来优化对这种独特的 ALS 亚型的治疗。

脊髓性肌萎缩症最佳实践更新
治愈 SMA(M. Schroth,JD),伊利诺伊州埃尔克格罗夫村;阿肯色大学医科学院神经内科儿科(KA),阿肯色州儿童医院,小石城;神经内科和神经肌肉护理中心(DC),德克萨斯州登顿;哥伦比亚大学欧文医学中心神经内科和儿科(DCDV),纽约;科罗拉多大学医学院儿科(MAG),奥罗拉;耶鲁大学医学院儿科(神经内科)(CI),康涅狄格州纽黑文;芝加哥 Ann & Robert H Lurie 儿童医院儿科和神经内科(NLK),伊利诺伊州西北范伯格医学院;路易斯维尔大学诺顿儿童医疗集团神经内科(AL);密歇根大学健康中心儿科(ENK),安娜堡;英国伦敦大奥蒙德街医院信托机构 Dubowitz 神经肌肉中心 (M. Scoto) 和英国伦敦大学学院大奥蒙德街儿童健康研究所;卡罗琳斯卡医学院妇女和儿童健康系 (TS)、卡罗琳斯卡大学医院儿童神经病学系、瑞典斯德哥尔摩阿斯特丽德林格伦儿童医院和香港新界沙田香港科学园神经肌肉骨骼修复医学中心;英国牛津大学 MDUK 牛津神经肌肉中心和 NIHR 牛津生物医学研究中心 (LS)、比利时列日大学儿科和列日大学医院神经肌肉中心;俄亥俄州辛辛那提儿童医院医疗中心和辛辛那提大学医学院儿科神经病学分部 (CT);基因治疗中心 (MAW)、阿比盖尔韦克斯纳研究所、全国儿童医院、儿科和神经病学部、俄亥俄州立大学韦克斯纳医学中心、哥伦布;以及运动神经元疾病科 (JFV-C)、拉菲医院、IIS La Fe、CIBERER、西班牙瓦伦西亚大学。
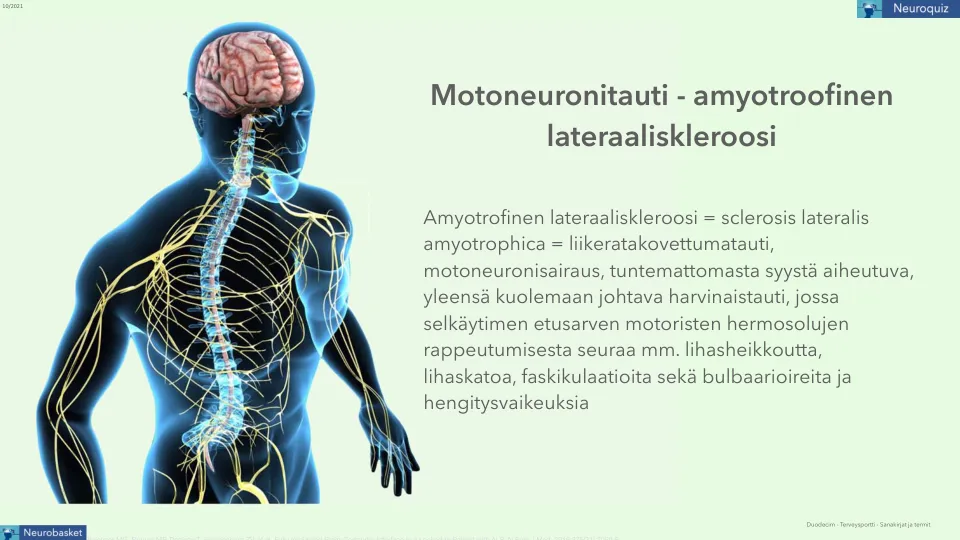
运动神经元疾病-肌萎缩侧索硬化症
肌萎缩侧索硬化症 = 肌萎缩侧索硬化症 = 运动神经元疾病,一种原因不明的罕见疾病,通常是致命的,脊髓前角运动神经元的退化会导致以下后果:肌肉无力、肌肉萎缩、肌束震颤以及延髓症状和呼吸困难

碱基编辑作为脊髓性肌萎缩症的基因治疗方法
图 1. 开发腺嘌呤碱基编辑来纠正 SMN2 外显子 7 C6T。a、未受影响个体和脊髓性肌萎缩症 (SMA) 患者的 SMN1 和 SMN2 示意图。SMN1 中的突变会导致 SMA,因为 SMN 蛋白会消耗,而这可以通过编辑 SMN2 来恢复。b、与 SMN1 相比,SMN2 外显子 7 C 到 T (C6T) 多态性的示意图,其中有碱基编辑器 gRNA 靶位及其估计的编辑窗口。cd、当使用由腺嘌呤脱氨酶结构域 ABEmax 33,38、ABE8.20m 35 和 ABE8e 36 与野生型 SpCas9(面板 c)或 SpRY 37(面板 d)融合的 ABE 时,对 SMN2 C6T 靶向腺嘌呤和其他旁观者碱基进行 A-to-G 编辑,通过靶向测序进行评估。 e,使用 SpRY 或其他宽松 SpCas9 PAM 变体 43 对 SMN2 外显子 7 中的腺嘌呤进行 A 到 G 编辑,通过靶向测序进行评估。图 ce 中的数据来自 HEK 293T 细胞中的实验;n = 3 个独立生物学重复的平均值、sem 和单个数据点。

肌萎缩侧索硬化症脑机接口的寿命
使用脑机接口 (BCI) 进行交流的持久性尚未得到广泛研究,这些接口适用于患有进行性神经退行性疾病的人。我们报告了一位患有晚期肌萎缩侧索硬化症 (ALS) 的患者 7 年独立在家使用植入式通信 BCI 的情况,该患者于 2016 年报告了开始使用该产品的情况。在家使用的频率随着时间的推移而增加,以替代逐渐失去对眼球注视追踪设备的控制,随后在植入 6 年后使用频率逐渐减少。当 BCI 控制变得不可靠时,在家使用就结束了。没有技术故障的迹象。相反,神经信号的幅度下降,计算机断层扫描成像显示进行性萎缩,这表明 ALS 相关的神经退行性病变最终导致 BCI 在多年成功使用后失效,尽管还有其他合理的解释。