XiaoMi-AI文件搜索系统
World File Search System荧光强度

Cas12a 针对多重基因组的优化
图 1. 现有 Cas12a CRISPRa 技术的评估。A) 采用两种不同的 Cas12a 核酸酶失活突变的 CRISPRa 构建体的比较。通过转导五天后表达 CD4 的细胞百分比来测量激活程度。B) 针对采用直接与 dCas12a (D908A) 连接的 TAD 组合的 12 种 CRISPRa 构建体变体,以基线表达为标准对 CD4 平均荧光强度 (MFI)。C) 示意图描绘了基于流式细胞术的平铺筛选的概览,该筛选用于识别其他活性 Cas12a CRISPRa 指南。D) 根据指南靶位点相对于 CD4、CD26、CD97 和 CD274 的转录起始位点 (TSS) 的位置绘制了每个指南在技术重复中的绝对最小 LFC 的 Z 分数。

IL-12工程技术的上级抗肿瘤活性(IOV- ... ) 基于T细胞的免疫疗法中的IOVANCE IP领导
方差分析,方差分析; Ctrl,控制; DP,药品; GSEA,基因集富集分析; GZMB,Granzyme B; EF-1α,伸长因子1α; HPAC,人类胰腺癌; ICO,可诱导的共刺激器; IFN-γ,干扰素伽玛; IL-2,白介素2; lag3,淋巴细胞活化基因3蛋白; MFI,平均荧光强度; NES,归一化富集评分; NFAT,活化T细胞的核因子; NS,不重要; PD-1,程序性细胞死亡蛋白1; REP,快速扩展方案; TCR,T细胞受体; TEIL-12,活化的T细胞 - 插鲁金12的膜束缚核因子; TIL,肿瘤浸润淋巴细胞; TIM3,T细胞免疫球蛋白结构域和粘蛋白结构域蛋白3; TME,肿瘤微环境; TNF-α,肿瘤坏死因子α。
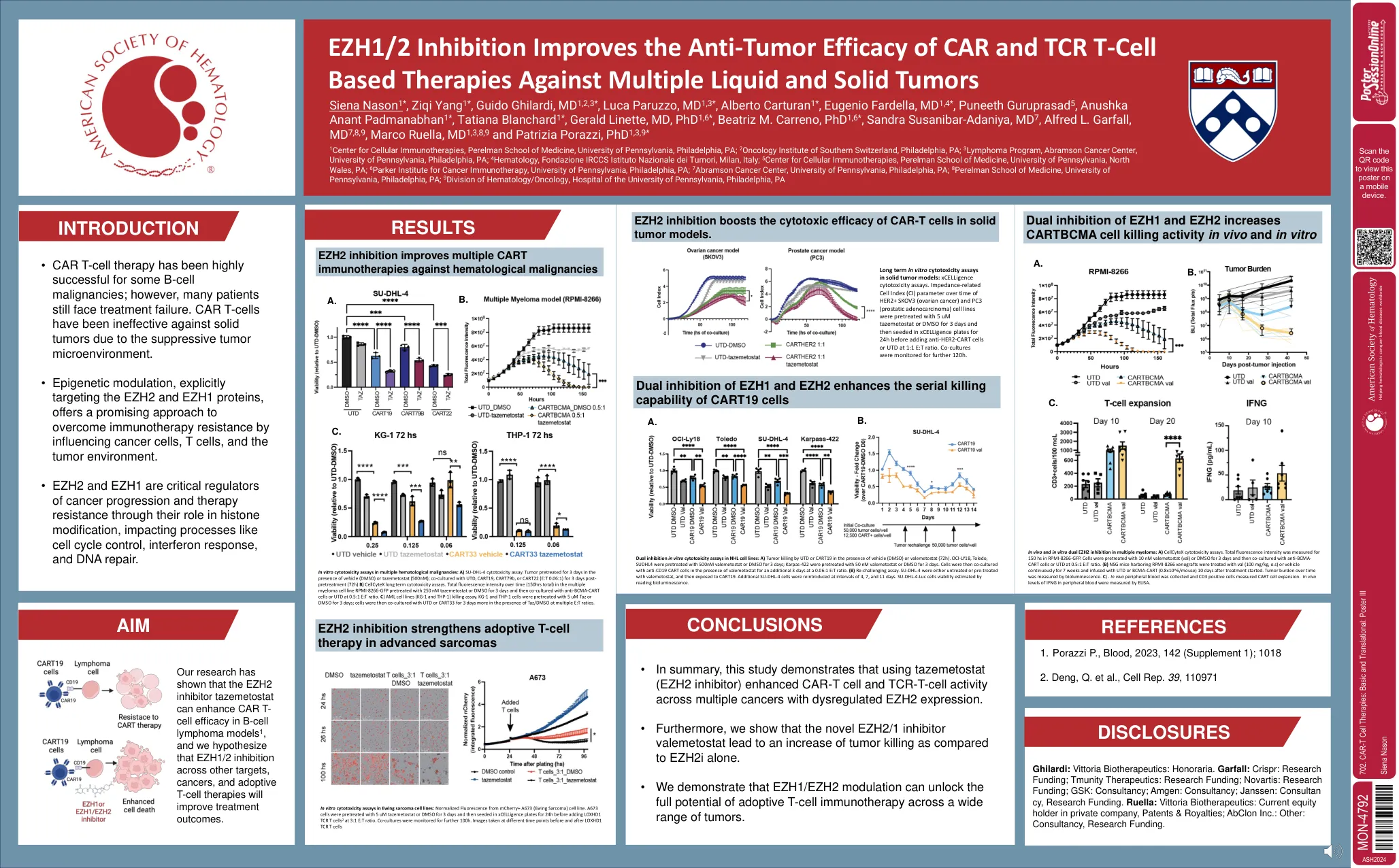
702。CAR-T细胞疗法:基本和翻译
在多种血液系统恶性肿瘤中的体外细胞毒性测定:a)SU-DHL-4细胞毒性测定。在存在媒介物(DMSO)或TazeMetostat(500nm)的情况下预处理3天;与UTD,CART19,CART79B或CART22(E:t 0.06:1)共同培养 - 预处理后3天(72h)b)CellcyTex长期细胞毒性测定。在多发性骨髓瘤细胞系RPMI-8266-GFP中,与250 nm tazemetostat或DMSO预处理3天的多发性骨髓瘤细胞系RPMI-8266-GFP中的总荧光强度3天,然后与抗BCMA-CART-CART-CART细胞或UTD在0.5:1 e:t的情况下共同培养。c)AML细胞系(KG-1和THP-1)杀死测定法。kg-1和Thp-1细胞用5 um taz或DMSO预处理3天;然后将细胞与UTD或CART33共培养3天,在多个E:T比的TAZ/DMSO存在下。

b"oxygen diffusion, creating an oxygen gradient throughout the coculture chamber. Basolateral gas flow containing 10% oxygen enters through the gas inlet , spreading evenly through the asymmetric coculture chamber with a magnetic stirrer . Exhaust gas is discharged through the gas outlet , completing the system's airflow (Fofanova et al., 2019). The figure was created using BioRender. (B) A physical picture of不对称的共培养室(C)在24小时内将FITC-二克的荧光强度添加到含有TIGK单层的Transwells的顶端腔室后的基底外侧。 \ XE2 \ X80 \ X9CNOROMOXIC \ XE2 \ X80 \ X9D与不对称培养条件下的差异化Tigks进行了比较(称为\ XE2 \ X80 \ x80 \ x9casymmmetric \ xe2 \ xe2 \ x80 \ x80 \ x9d)。在切换到含有Ca 2+的分化培养基之前,未分化的TIGK单层在正常氧条件下培养。 (N.S。:P> 0.05,***:P <0.001,n = 2技术重复,n = 3个生物重复序列)。 (d)在固定培养培养基培养基培养基中,在常氧培养基中维持的Transwell插入物中的Tigk单层的形态或在非对称共培养室中培养的24小时。已知胶原蛋白会影响明亮的田间成像
b“氧扩散,在整个共培养室中产生氧梯度。含有10%氧气的基底外侧气流通过气体入口进入,并用磁性搅拌器均匀地通过不对称的共培养室扩散。排气通过气体插座排放,完成了系统的气流(Fofanova等,2019)。该图是使用生物者创建的。(b)不对称共培养室的物理图片。(c)在将FITC-DEXTRAN添加到包含Tigk单层的Transwells的顶端室后,在24小时内比较了基底外侧室内FITC-脱骨的荧光强度。在常规氧培养条件下未分化(阴性对照)和分化的Tigks(称为\ XE2 \ X80 \ X9CNORMOXIC \ XE2 \ X80 \ X9D)与在不对称培养条件下的分化Tigk(称为AS AS AS) \ xe2 \ x80 \ x9casymmetric \ xe2 \ x80 \ x9d)。对于每种条件,减去空白培养基的背景荧光强度。未分化的TIGK单层在正常氧状态下培养,然后切换为包含Ca 2+的分化培养基,用作负面对照。(N.S.:p> 0.05,***:p <0.001,n = 2技术重复,n = 3个生物重复序列)。(e)在常氧和不对称培养条件下培养的TIGK单层中细胞活力的比较。热处理细胞是阴性对照(N.S.:p> 0.05,**:p <0.01,n = 3,n = 3)。(d)Transwell插入物中的Tigk单层的形态在正常氧化条件下维持在细胞培养培养基中,或在不对称的共培养室中培养24小时。已知胶原蛋白由于胶原纤维的存在而影响明亮的田间成像,与未涂层的表面相比,该胶原纤维可能会掩盖所观察到的细胞或结构的细节(Hashimoto等,2020)。

Quantaurus-Tau荧光寿命光谱仪C16361系列
从有机材料或荧光探针中获得的荧光光谱是控制和评估材料功能和特性的重要参数,例如峰值波长和荧光强度。但是,荧光光谱通常显示时间整合的信息,因此,当材料包含多种物质和反应性元素时,它们的荧光光谱只能作为集成信息获取。在这种情况下,一种有效的方法是通过使用时轴参数来观察光发射动力学。这通常称为荧光寿命测量,其中通过脉冲光激发的物质返回其基态所需的时间是在亚纳秒到毫秒到毫秒的区域中测量的。此测量允许获得更多信息,例如在相同的波长和材料中存在的百分比等多种不同的荧光寿命等。

肿瘤衍生的仿生纳米元素具有免疫逃避能力,可增强低剂量放射疗法
在4T1肿瘤细胞中,CF和RF的溶血跟踪器绿色FM(蓝色)和DIL(红色)共定位。(b)使用ImageJ软件确定的(a)的DIL荧光强度。(c)JC-1(JC-1单体绿色,在不同处理下用于JC-1的荧光图像红色。(d)使用DAPI和-H2AX染色在所示的细胞中使用DAPI和-H2AX染色可视化核凝结和DNA碎片,并显示了代表性的图片。(e)基于每个处理组100个细胞(γ-H2AX焦点/100μm2,n = 3)的分析,确定了γ-H2AX灶的密度。(f)使用用2或6 Gy辐射处理的4T1细胞(n = 3)进行了菌落形成测定。(g)PMSI对细胞内的影响

1个国家主要实验室,微生物学院,中国科学院,北京100101,中国 *通信:liwei_zhou1982@im。2江南大学的未来食品科学中心,中国214122 *通信:yanfengliu@jiangnan.edu.cn收到:2024年1月30日; AC
图1。在大肠杆菌的正交线性质粒(O-Replicon)中的正交复制系统的结构和表征不干扰宿主基因组的复制,在大肠杆菌中已成功发展。末端蛋白(TP),正交DNAP(O-DNAP),双链DNA结合蛋白(DSB)和PRD1噬菌体的单链DNA结合蛋白(SSB)的基因在iPTG-诱导促销基因组合的控制下被编码并表达。这些蛋白质与含有倒末端重复序列的异源基因的DNA序列(ITR)相互作用,最终在体内获得正交复制线性质粒。此外,戈伊斯可以通过设计的 - 毒素O-DNAP的连续作用来实现快速而独立的进化,例如在短时间内的短期内显着提高细胞对抗生素的耐药性以及GFP的荧光强度。

材料进步-RSC出版
缓慢的响应率和低分辨率是其他缺点。5另一方面,基于磷的发光的光学测量法可以更好地相对温度敏感性,较短的获取时间,空间分辨率等。6–8用于光学温度测量法的磷光体被称为热磷,它们具有依赖温度的发光参数,例如发射强度,衰减或上升时间,发射颜色和光谱变化。在这些方法中,基于荧光强度比(FIR)的温度传感可通过可忽略不计的漂移和自我引用来更好地传感。9–12关于FIR方法的大多数报告都集中在稀土离子的热耦合水平(TCLS)上。由于20 o d e o 2000 cm 1的较小能量隙引起的重叠发射限制了传感器的准确性和信号可区分性。13–16因此,基于非TCL的FIR引起了很大的关注。在这种情况下,涉及双重发射限制的声子辅助的能量转移有助于实现更好的相对温度灵敏度和信号可区分性。17,18

大分子拥挤在生物打印的人横纹肌肉瘤模型中调整了细胞外基质沉积
MMC对RH30和RD球体的影响。 a如果在Rh30 -arms-(左)(左)和RD -erms-(右)球体上染色(FN; Green)和胶原I(大肠杆菌;红色),则在不存在MMC处理的情况下冷冻切片(DAPI,cell核,蓝色),比例尺=50μm。 B胶原蛋白I的平均荧光强度(MFI)和球体冷冻切片中的纤连蛋白表达。 c离开。 在所有测试条件下播种在ULA板中的球体的相对形图像,以及井底RH30粘附细胞的细节。 比例尺=右200μm。 如果在RH30和RD球体中用MMC处理的纤连蛋白和胶原蛋白I的染色显示RH30球体下方的粘附细胞的存在。 比例尺=50μm。 d无需MMC处理的RH30和RD球体的形状参数(面积,周长,圆度和坚固),n = 12,Student t -test*p <0.05,** p <0.01,**** p <0.0001。 (为了解释该图传奇中对颜色的引用,读者被转介给本文的网络版本。)MMC对RH30和RD球体的影响。a如果在Rh30 -arms-(左)(左)和RD -erms-(右)球体上染色(FN; Green)和胶原I(大肠杆菌;红色),则在不存在MMC处理的情况下冷冻切片(DAPI,cell核,蓝色),比例尺=50μm。 B胶原蛋白I的平均荧光强度(MFI)和球体冷冻切片中的纤连蛋白表达。c离开。在所有测试条件下播种在ULA板中的球体的相对形图像,以及井底RH30粘附细胞的细节。比例尺=右200μm。如果在RH30和RD球体中用MMC处理的纤连蛋白和胶原蛋白I的染色显示RH30球体下方的粘附细胞的存在。比例尺=50μm。 d无需MMC处理的RH30和RD球体的形状参数(面积,周长,圆度和坚固),n = 12,Student t -test*p <0.05,** p <0.01,**** p <0.0001。(为了解释该图传奇中对颜色的引用,读者被转介给本文的网络版本。)

通过转录重编程发现抗癌药物的高通量策略
转录重编程是癌症进展和复发的因素,然而肿瘤转录重编程机制不甚明了,导致有效药物的研发十分困难,而基因表达特征有助于将遗传信息与药物治疗联系起来。到目前为止,基于基因表达特征的高通量药物发现方法有两种:L1000(测量 978 个“标志性”基因的 mRNA 转录本丰度)和基于高通量测序的高通量筛选(HTS 2),适用于针对转录重编程的抗癌药物发现。L1000 利用连接介导的扩增和与 Luminex 珠子的杂交,通过检测珠子颜色和藻红蛋白信号的荧光强度来突出显示基因表达变化。HTS 2 利用 RNA 介导的寡核苷酸退火、选择和连接以及高通量测序,通过直接测量基因序列来量化基因表达变化。本文综述了L1000和HTS 2 的技术原理和应用,并讨论了它们在抗癌药物研发中的优势和局限性。